

The RASAL2 variant promotes aberrant RAS signaling and resistance to anti-EGFR therapy in colorectal cancer
- PMID: 40849341
- PMCID: PMC12375080
- DOI: 10.1038/s41598-025-16325-6
The RASAL2 variant promotes aberrant RAS signaling and resistance to anti-EGFR therapy in colorectal cancer
Abstract
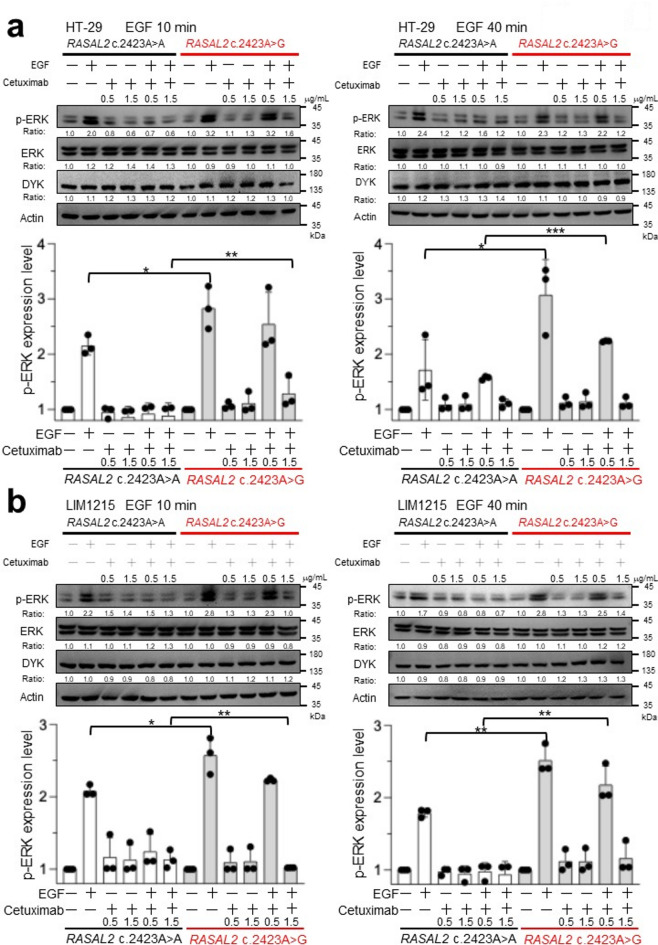
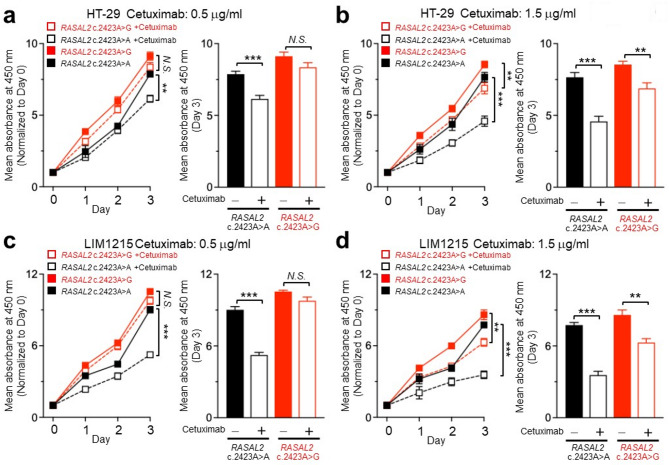
Anti-EGFR monoclonal antibodies are essential for metastatic colorectal cancer (CRC) treatment, however, resistance remains problematic in KRAS/NRAS/BRAF wild-type patients. RAS protein activator-like 2 (RASAL2) regulates RAS signaling by catalyzing the conversion of RAS. This study investigates the pathogenicity of the germline RASAL2 c.2423 A > G variant, identified in a high-risk family, and its potential role in CRC progression and therapy resistance. Population analysis reveals its rarity in East Asians (0.01%) but an increased prevalence in Taiwanese CRC patients (1.63%). Functional studies demonstrate that RASAL2 c.2423 A > G enhances RAS signaling, causing sustained ERK phosphorylation and increased CRC cell proliferation. Additionally, RASAL2-mutant cells require higher doses of cetuximab for ERK suppression and growth inhibition, indicating resistance to anti-EGFR therapy via abnormal RAS activation. According to the American College of Medical Genetics and Genomics criteria, the variant is likely pathogenic. Our study highlights RASAL2 c.2423 A > G as a potential biomarker for CRC risk and therapy response.
Keywords: RASAL2; Anti-EGFR; Colorectal cancer; Germline variant; Pathogenic; RAS signaling; Resistance.
© 2025. The Author(s).
Conflict of interest statement
Declarations. Competing interests: The authors declare no competing interests. Running title: RASAL2 variant mediates anti-EGFR resistance in colorectal cancer. Word count: 3068, 6 figures. Competing Interests: The authors declare no conflicts of interest. Figures and Figure Legends.
Figures




Similar articles
-
The predictive value of KRAS, NRAS, BRAF, PIK3CA and PTEN for anti-EGFR treatment in metastatic colorectal cancer: A systematic review and meta-analysis.Acta Oncol. 2014 Jul;53(7):852-64. doi: 10.3109/0284186X.2014.895036. Epub 2014 Mar 25. Acta Oncol. 2014. PMID: 24666267
-
Epidermal growth factor receptor (EGFR) inhibitors for metastatic colorectal cancer.Cochrane Database Syst Rev. 2017 Jun 27;6(6):CD007047. doi: 10.1002/14651858.CD007047.pub2. Cochrane Database Syst Rev. 2017. PMID: 28654140 Free PMC article.
-
EGFR gene gain and PTEN protein expression are favorable prognostic factors in patients with KRAS wild-type metastatic colorectal cancer treated with cetuximab.J Cancer Res Clin Oncol. 2014 May;140(5):737-48. doi: 10.1007/s00432-014-1626-2. Epub 2014 Mar 5. J Cancer Res Clin Oncol. 2014. PMID: 24595598 Free PMC article.
-
KRAS p.G13D mutations are associated with sensitivity to anti-EGFR antibody treatment in colorectal cancer cell lines.J Cancer Res Clin Oncol. 2013 Feb;139(2):201-9. doi: 10.1007/s00432-012-1319-7. Epub 2012 Sep 27. J Cancer Res Clin Oncol. 2013. PMID: 23015072 Free PMC article.
-
ALKBH5 suppresses m6A mRNA modification of FOXM1 to drive Cetuximab resistance in KRAS-mutant colorectal cancer.Oncogene. 2025 Sep;44(35):3225-3238. doi: 10.1038/s41388-025-03490-1. Epub 2025 Jul 7. Oncogene. 2025. PMID: 40624314
References
-
- Van Cutsem, E. et al. Cetuximab and chemotherapy as initial treatment for metastatic colorectal cancer. N Engl. J. Med.360, 1408–1417 (2009). - PubMed
-
- Douillard, J. Y. et al. Randomized, phase III trial of panitumumab with infusional fluorouracil, leucovorin, and oxaliplatin (FOLFOX4) versus FOLFOX4 alone as first-line treatment in patients with previously untreated metastatic colorectal cancer: the PRIME study. J. Clin. Oncol.28, 4697–4705 (2010). - PubMed
-
- Perrone, F. et al. PI3KCA/PTEN deregulation contributes to impaired responses to cetuximab in metastatic colorectal cancer patients. Ann. Oncol.20, 84–90 (2009). - PubMed
-
- Sartore-Bianchi, A. et al. PIK3CA mutations in colorectal cancer are associated with clinical resistance to EGFR-targeted monoclonal antibodies. Cancer Res.69, 1851–1857 (2009). - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
