

Multi-omics analysis identifies an M-MDSC-like immunosuppressive phenotype in lineage-switched AML with KMT2A rearrangement
- PMID: 40858576
- PMCID: PMC12381044
- DOI: 10.1038/s41467-025-63271-y
Multi-omics analysis identifies an M-MDSC-like immunosuppressive phenotype in lineage-switched AML with KMT2A rearrangement
Abstract
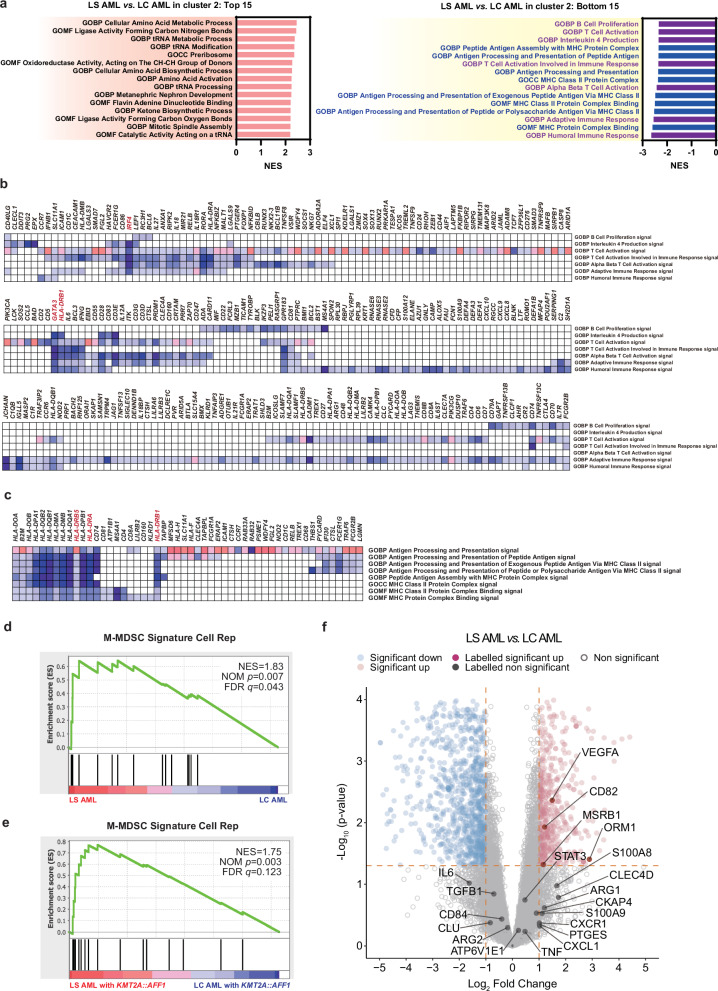
Lineage switching (LS) is the conversion of cancer cell lineage during the course of a disease. LS in leukemia cell lineage facilitates cancer cells escaping targeting strategy like CD19 targeted immunotherapy. However, the genetic and biological mechanisms underlying immune evasion by LS leukemia cells are not well understood. Here, we conduct a multi-omics analysis of patient samples and find that lineage-switched acute myeloid leukemia (LS AML) cells with KMT2A rearrangement (KMT2A-r) possess monocytic myeloid derived suppressor cell (M-MDSC)-like characteristics. Single-cell mass cytometry analysis reveals an increase in the M-MDSC like LS AML as compared to those of lineage-consistent KMT2A-r AML, and single-cell transcriptomics identify distinct expression patterns of immunoregulatory genes within this population. Furthermore, in vitro assays confirm the immunosuppressive capacity of LS AML cells against T cells, which is analogous to that of MDSCs. These data provide insight into the immunological aspects of the complex pathogenesis of LS AML, as well as development of future treatments.
© 2025. The Author(s).
Conflict of interest statement
Competing interests: The authors declare no competing interests.
Figures




References
-
- Rossi, J. G. et al. Lineage switch in childhood acute leukemia: an unusual event with poor outcome. Am. J. Hematol.87, 890–897 (2012). - PubMed
-
- Kurzer, J. H. & Weinberg, O. K. To B- or not to B-: a review of lineage switched acute leukemia. Int. J. Lab Hematol.44, 64–70 (2022). - PubMed
-
- Silbert, S. K. et al. Project evolve, evaluation of lineage switch (LS), an International Initiative: preliminary results reveal dismal outcomes in patients with LS. Blood142, 4202–4202 (2023).
MeSH terms
Substances
Grants and funding
- JP24ck0106935/Japan Agency for Medical Research and Development (AMED)
- 22ama221514h0001/Japan Agency for Medical Research and Development (AMED)
- 23ama221514h002/Japan Agency for Medical Research and Development (AMED)
- JP22cm0106xxxh000x/Japan Agency for Medical Research and Development (AMED)
- 23ama221505h0002/Japan Agency for Medical Research and Development (AMED)
LinkOut - more resources
Full Text Sources
Medical
