

Genome sequencing provides high diagnostic yield and new etiological insights for intellectual disability and developmental delay
- PMID: 40858643
- PMCID: PMC12381280
- DOI: 10.1038/s41525-025-00521-4
Genome sequencing provides high diagnostic yield and new etiological insights for intellectual disability and developmental delay
Abstract
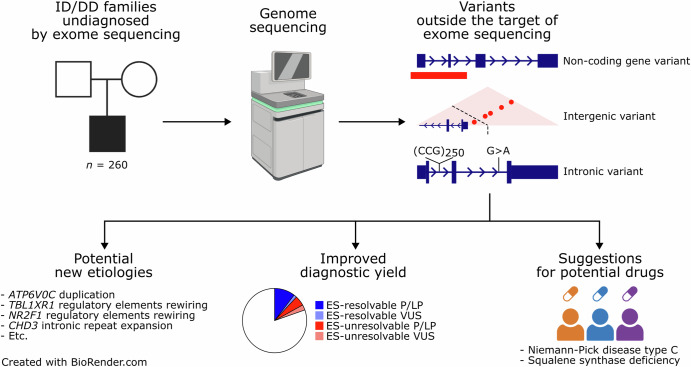
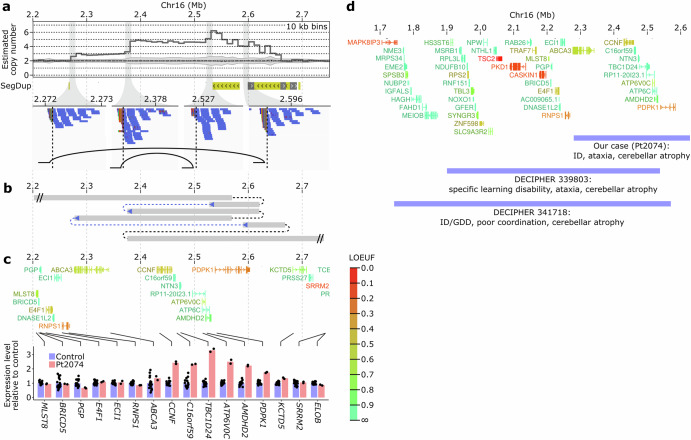
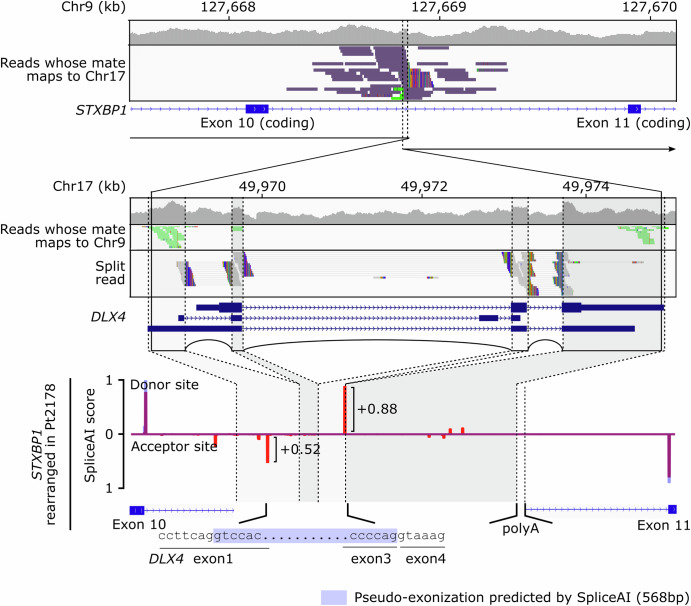
Short-read genome sequencing (GS) is a powerful technique for investigating the genetic etiologies of rare diseases, capturing diverse genetic variations that are challenging to approach with exome sequencing (ES). We performed GS on 260 families with intellectual disability/developmental delay. GS detected potentially disease-related variants in 55 of the 260 families, with structural resolution by long-read sequencing or optical genome mapping, and functional assessment by RNA sequencing. Excluding 31 theoretically ES-resolvable cases, GS yielded likely pathogenic variants in 17 of 229 as well as variants of unknown significance in 7 of 229, totaling 10.5%. These variants implicated several new etiological mechanisms: a microduplication syndrome involving ATP6V0C; disturbed interactions of TBL1XR1 and NR2F1 with putative cis-regulatory elements by chromosomal rearrangements; and a CCG repeat expansion near the CHD3 transcription start site. This study highlights the critical role of GS in clinical diagnostics and its potential to advance understanding of genetic disorders.
© 2025. The Author(s).
Conflict of interest statement
Competing interests: The authors declare no competing interests.
Figures



References
-
- Lindstrand, A. et al. Genome sequencing is a sensitive first-line test to diagnose individuals with intellectual disability. Genet. Med.24, 2296–2307 (2022). - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
