

Astrocyte-neuron combined targeting for CYP46A1 gene therapy in Huntington's disease
- PMID: 40859407
- PMCID: PMC12382279
- DOI: 10.1186/s40478-025-02054-4
Astrocyte-neuron combined targeting for CYP46A1 gene therapy in Huntington's disease
Abstract
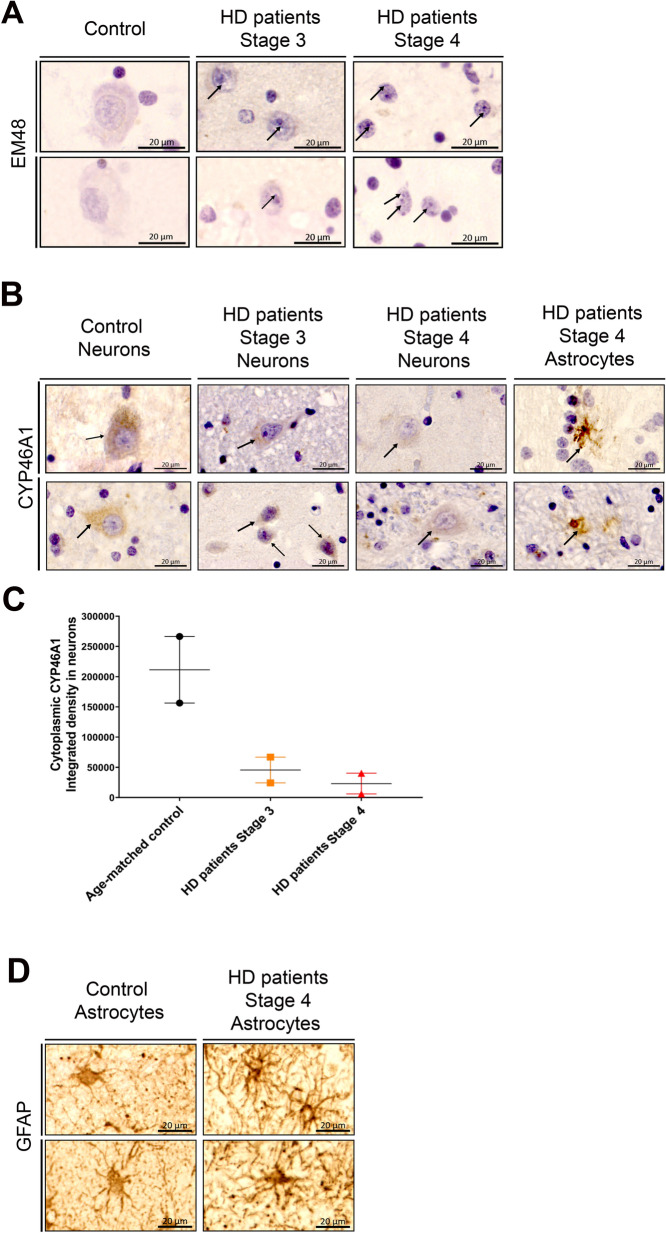
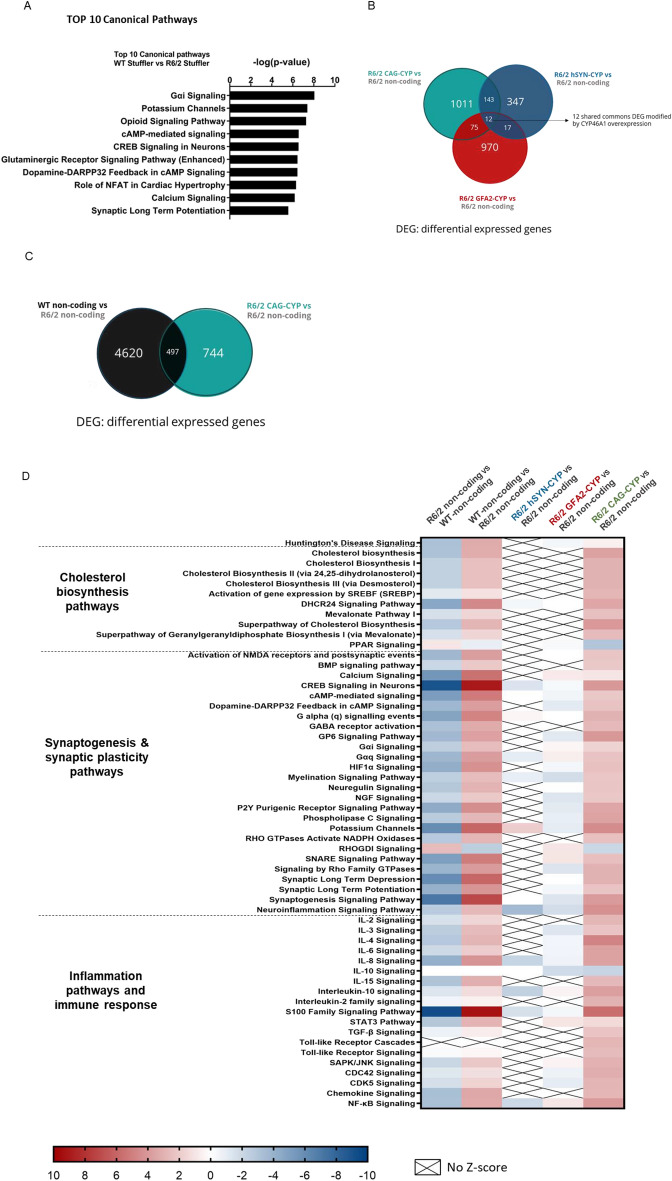
Huntington's disease (HD) is an autosomal dominant neurodegenerative disease caused by an abnormal expansion of cytosine-adenine-guanosine (CAG) trinucleotidein the huntingtin gene. Mutant huntingtin (mHTT) expression in neurons and glial cells affects neuron and astrocyte functions and leads to the loss of medium spiny neurons of the striatum. Brain cholesterol pathway is severely affected by HTT mutation in neurons and astrocytes, contributing to HD pathogenesis. Decreased cholesterol production and transport by astrocytes impair synapse maturation and neurotransmission. Brain cholesterol metabolism is maintained by cholesterol hydroxylation into 24-hydroxycholesterol by the neuronal enzyme cholesterol 24-hydroxylase (CYP46A1). CYP46A1 is decreased in affected brain regions in HD patients and mice. AAV-CYP46A1 striatal delivery was shown to restore cholesterol metabolism with neuroprotective effects in two mouse models of HD, characterized by mHTT aggregates' reduction, improved transcriptomic profile, and Brain-Derived Neurotrophic Factor (BDNF) signaling, and preservation of striatal neurons. From a therapeutic perspective, we intended to clarify the detailed mechanisms and the specific role of neurons and astrocytes in the therapeutic effects of AAV-CYP46A1 delivery. We first evaluated CYP46A1 expression in astrocytes in HD post-mortem putamen at a late stage of disease progression. To determine the specific contribution of CYP46A1 expression in astrocytes compared to neurons on the HD phenotype, we assessed the effects of AAV-CYP46A1 striatal injection under the control of astrocytic (GFA2) or neuronal (hSYN) promoters in R6/2 mice. Overall, equivalent transgenic CYP46A1 protein levels, both astrocytic and neuronal targeting, mitigate medium ppiny neuron (MSN) atrophy and improve spine density in R6/2 mice. Reduction of mHTT aggregates in neurons is similar when CYP46A1 is overexpressed in neurons or in astrocytes. However, astrocyte targeting reduces mHTT aggregates in neurons and astrocytes, while restricted neuronal targeting reduces mHTT aggregates in neurons only. Altogether, astrocytic targeting of CYP46A1 expression in CYP46A1-tested animals combines cell-autonomous and non-cell-autonomous mechanisms of action, with improved phenotypic correction compared to neuronal-restricted targeting. Allowing expression in both cell types with higher expression levels of CYP46A1 showed overall better efficacy. We demonstrate that astrocyte-neuron combined targeting with AAV-CAG-CYP46A1 delivery increases therapeutic efficacy. This study brings new evidence that CAG-mediated CYP46A1 striatal overexpression significantly modifies the transcriptome in R6/2 mice for pathways involved in synaptogenesis and inflammation, suggesting targeting both astrocytes and neurons provides benefits for HD phenotypic correction.
Keywords: Astrocytes; Cholesterol; Inflammation; Neurons; Synapses.
© 2025. The Author(s).
Conflict of interest statement
Declarations. Ethics approval consent to participate: Regarding experiments realized on R6/2 mice and WT-littermates, all animal procedures and experiments were approved by the local ethical committee (Ethical Committee of IBPS, n° 05) and the French Ministry of National Education, Higher Education and Research (reference number APAFIS #12412–2017113010191653 v7) and were performed in accordance with the Guide for the Care and Use of Laboratory Animals (US National Institutes of Health). The HD brains were donated after informed consent by the patients themselves during life and these donations were supported by the spouse and doctors of these patients. The Medical Ethical committee of the University Medical Centre Groningen, the Netherlands approved the brain donation procedure, as well as the donation form and patient information. Consent for publication: I give my consent for the publication of the manuscript titled Astrocyte-neuron combined targeting for CYP46A1 gene therapy in Huntington’s disease in Acta Neuropathologica communications. All co-authors have approved the final version, and we agree to the submission and publication. I confirm that all necessary permissions and consents (including for images or personal data) have been obtained. Competing interests: The clinical application of gene therapy approach for CYP46A1 delivery in Huntington’s disease is protected by the published patent filed in Europe and the United States, PCT/EP2011/068033 and WO2012049314A1, respectively.
Figures





References
-
- Bates GP, Dorsey R, Gusella JF et al (2015) Huntington disease. Nat Rev Dis Primers 1(April):1–21. 10.1038/nrdp.2015.5 - PubMed
-
- Ghosh R, Tabrizi SJ (2018) Huntington disease. Handbook of Clin Neurol 147:255–278. 10.1016/B978-0-444-63233-3.00017-8 - PubMed
-
- MacDonald ME, Ambrose CM, Duyao MP et al (1993) A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. Cell 72(6):971–983. 10.1016/0092-8674(93)90585-E - PubMed
-
- Saudou F, Humbert S (2016) The Biology of Huntingtin. Neuron 89(5):910–926. 10.1016/j.neuron.2016.02.003 - PubMed
-
- Roze E, Saudou F, Caboche J (2008) Pathophysiology of Huntington’s disease: from huntingtin functions to potential treatments. Curr Opin Neurol 21(4):497–503. 10.1097/WCO.0b013e328304b692 - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
