

Decoupling transcriptome layers: the distinct and variable nature of circular RNAs
- PMID: 40866873
- PMCID: PMC12392627
- DOI: 10.1186/s12915-025-02375-9
Decoupling transcriptome layers: the distinct and variable nature of circular RNAs
Abstract
Background: Circular RNAs (circRNAs) and mRNAs are distinct transcripts from the same genes, produced by different splicing mechanisms. This study investigates the behavior of the circular transcriptome relative to the linear one across biological conditions and tissues. We analyzed transcriptomic data from 36 bovine monocyte-derived macrophage (MDM) samples collected during an ex vivo Mycobacterium avium ssp. paratuberculosis (MAP) infection experiment, stratified by Johne's disease (JD) antibody status (JD+ or JD-) and by infection condition (control or MAP infected). We extended our analysis to healthy bovine tissues, including neonatal and post-pubertal testes, and liver and muscle samples from 12 animals stratified by sex and feed efficiency.
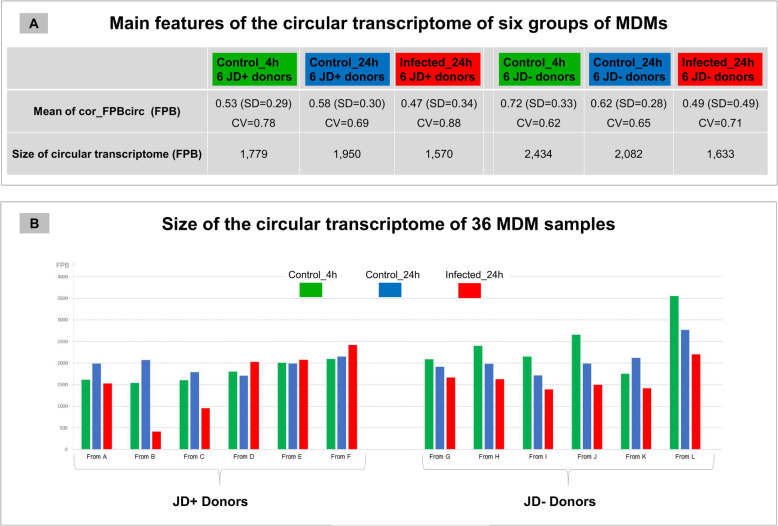
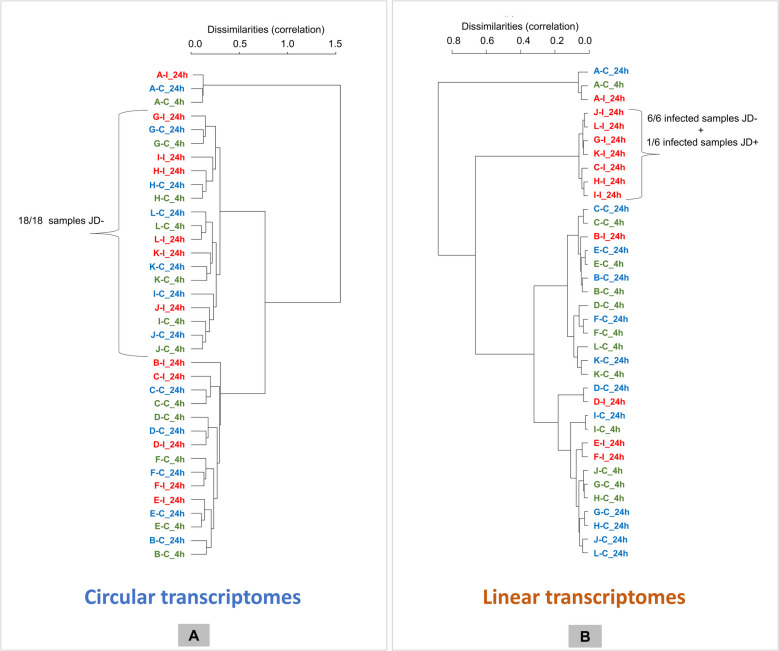
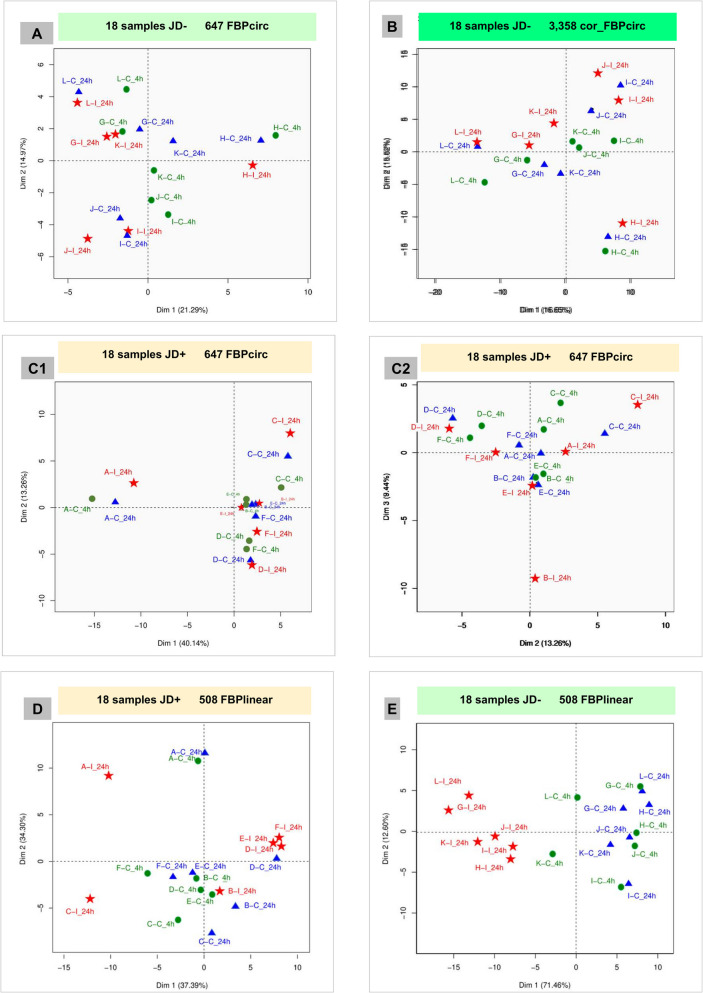
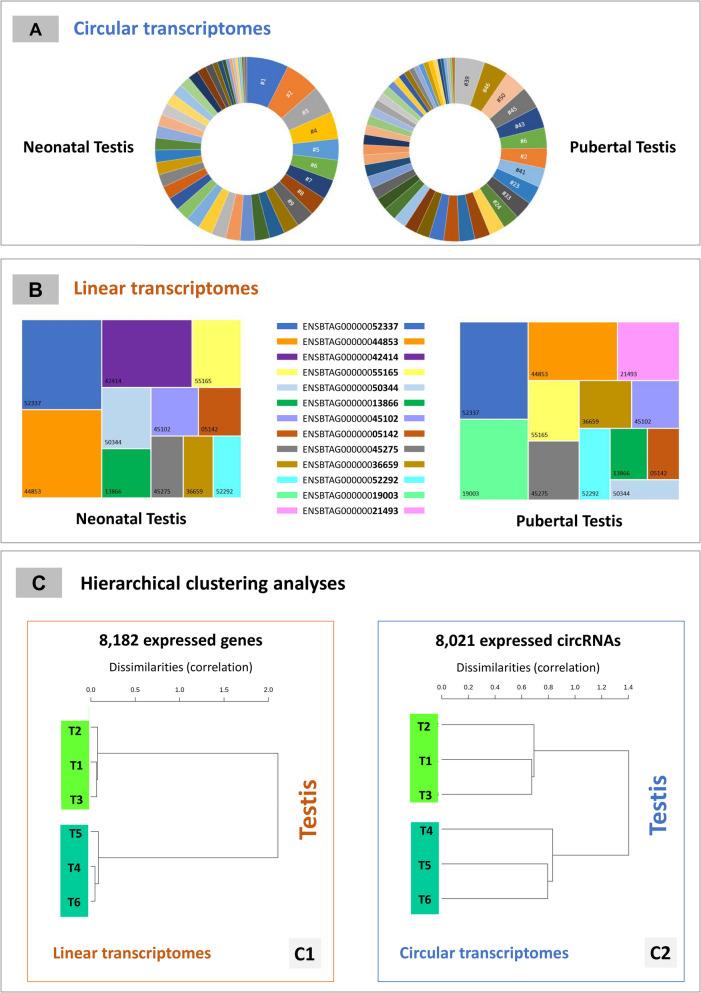
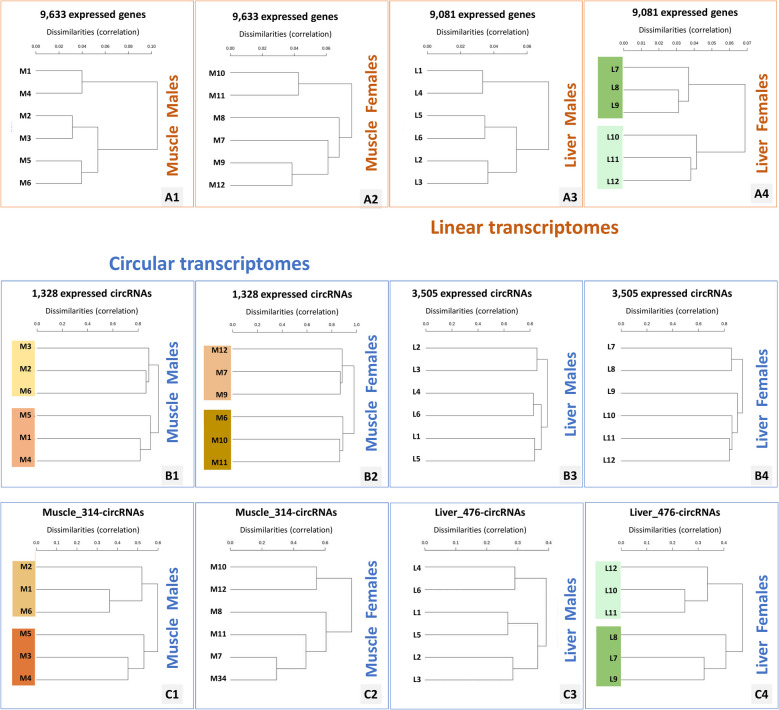
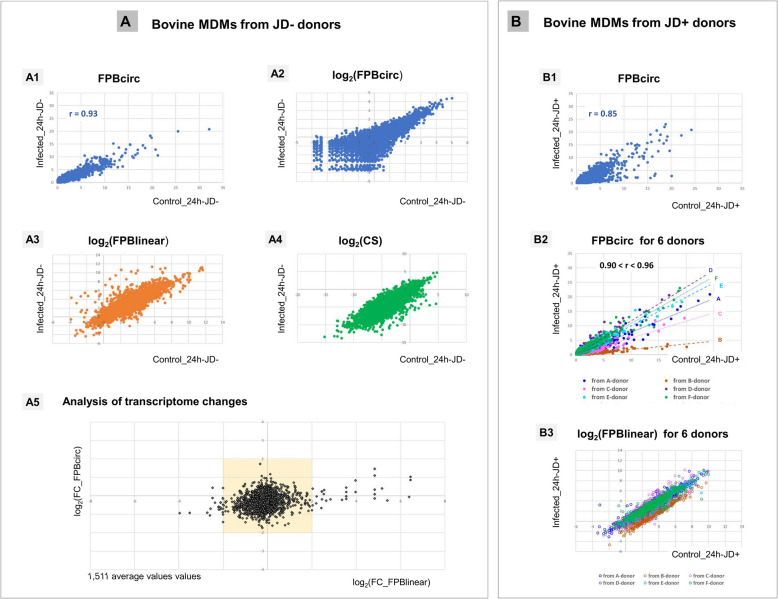
Results: In the 36 MDM samples, we identified 3358 exonic circRNAs derived from 1895 genes. By comparing the mean expression levels of circRNAs and linear transcripts, and considering the number of expressed genes, we estimate that the circular transcriptome is approximately 100 times smaller than the linear transcriptome. Analyses of the circular and linear transcriptomes revealed that MAP infection impacted only the linear transcriptome of MDM_JD- . The other three transcriptomes-circular JD- , circular JD+ , and linear JD+ -showed no infection-specific response. In the testes, maturation was associated with profound but uncoordinated changes in the circular and linear transcriptomes. While circRNA abundance declined, the linear transcriptome underwent a complete reorganization marked by the activation of novel genes. In the liver, female samples clustered by feed efficiency only when the entire linear and top-expressed circular transcriptomes were considered, respectively. In MDMs, the circular transcriptomes of control and infected samples, as well as the JD+ linear transcriptome, were dominated by donor-specific signatures. In contrast, the JD- linear transcriptome reflected MAP infection, with infection-specific structuring overriding inter-individual variation.
Conclusions: In both MDM and tissue samples, circular and linear transcriptomes follow distinct and largely independent regulatory logics. While both capture inter-individual variation, circRNA expression appears more variable and may carry fewer physiological signals, especially when no clear phenotypic signature has been detected in the corresponding linear transcriptome. These findings demonstrate that circular and linear RNAs arise from complementary and nonredundant layers of gene regulation, emphasizing the importance of analyzing both in parallel.
Keywords: Circular transcriptome; Exonic circRNAs; Expression; Linear transcriptome; Parental genes.
© 2025. The Author(s).
Conflict of interest statement
Declarations. Competing interests: The authors declare no competing interests.
Figures

References
-
- Kristensen LS, Andersen MS, Stagsted LVW, Ebbesen KK, Hansen TB, Kjems J. The biogenesis, biology and characterization of circular RNAs. Nat Rev Genet. 2019;20:675–91. - PubMed
-
- Zhang J, Zhao F. Circular RNA discovery with emerging sequencing and deep learning technologies. Nat Genet. 2025;57(5):1089–102. - PubMed
-
- Robic A, Kühn C. Circular RNA and backsplicing: unraveling the real, the misconceptions, and the unknown. Genomics Commun. 2025;2: e004.
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
