

Type 2 Diabetes and the Multifaceted Gut-X Axes
- PMID: 40871736
- PMCID: PMC12389143
- DOI: 10.3390/nu17162708
Type 2 Diabetes and the Multifaceted Gut-X Axes
Abstract
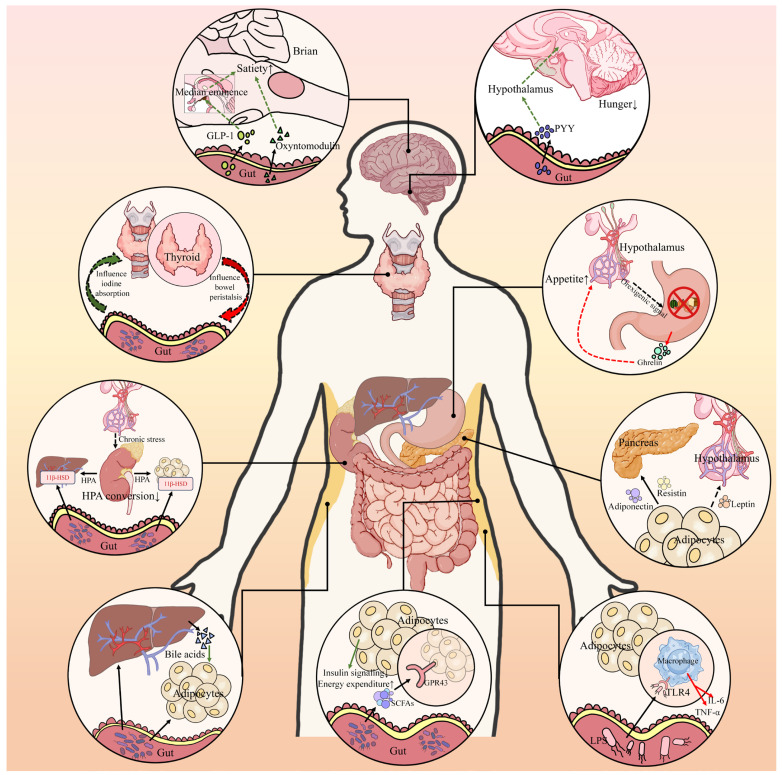
Type 2 diabetes (T2D) is a complex metabolic disease characterized by chronic hyperglycemia due to insulin resistance and inadequate insulin secretion. Beyond the classically implicated organs, emerging evidence highlights the gut as a central player in T2D pathophysiology through its interactions with metabolic organs. The gut hosts trillions of microbes and enteroendocrine cells that influence inflammation, energy homeostasis, and hormone regulation. Disruptions in gut homeostasis (dysbiosis and increased permeability) have been linked to obesity, insulin resistance, and β-cell dysfunction, suggesting multifaceted "Gut-X axes" contribute to T2D development. We aimed to comprehensively review the evidence for gut-mediated crosstalk with the pancreas, endocrine system, liver, and kidneys in T2D. Key molecular mechanisms (incretins, bile acids, short-chain fatty acids, endotoxins, etc.) were examined to construct an integrated model of how gut-derived signals modulate metabolic and inflammatory pathways across organs. We also discuss clinical implications of targeting Gut-X axes and identify knowledge gaps and future research directions. A literature search (2015-2025) was conducted in PubMed, Scopus, and Web of Science, following PRISMA guidelines (Preferred Reporting Items for Systematic Reviews). Over 150 high-impact publications (original research and review articles from Nature, Cell, Gut, Diabetologia, Lancet Diabetes & Endocrinology, etc.) were screened. Data on gut microbiota, enteroendocrine hormones, inflammatory mediators, and organ-specific outcomes in T2D were extracted. The GRADE framework was used informally to prioritize high-quality evidence (e.g., human trials and meta-analyses) in formulating conclusions. T2D involves perturbations in multiple Gut-X axes. This review first outlines gut homeostasis and T2D pathogenesis, then dissects each axis: (1) Gut-Pancreas Axis: how incretin hormones (GLP-1 and GIP) and microbial metabolites affect insulin/glucagon secretion and β-cell health; (2) Gut-Endocrine Axis: enteroendocrine signals (e.g., PYY and ghrelin) and neural pathways that link the gut with appetite regulation, adipose tissue, and systemic metabolism; (3) Gut-Liver Axis: the role of microbiota-modified bile acids (FXR/TGR5 pathways) and bacterial endotoxins in non-alcoholic fatty liver disease (NAFLD) and hepatic insulin resistance; (4) Gut-Kidney Axis: how gut-derived toxins and nutrient handling intersect with diabetic kidney disease and how incretin-based and SGLT2 inhibitor therapies leverage gut-kidney communication. Shared mechanisms (microbial SCFAs improving insulin sensitivity, LPS driving inflammation via TLR4, and aryl hydrocarbon receptor ligands modulating immunity) are synthesized into a unified model. An integrated understanding of Gut-X axes reveals new opportunities for treating and preventing T2D. Modulating the gut microbiome and its metabolites (through diet, pharmaceuticals, or microbiota therapies) can improve glycemic control and ameliorate complications by simultaneously influencing pancreatic islet function, hepatic metabolism, and systemic inflammation. However, translating these insights into clinical practice requires addressing gaps with robust human studies. This review provides a state-of-the-art synthesis for researchers and clinicians, underlining the gut as a nexus for multi-organ metabolic regulation in T2D and a fertile target for next-generation therapies.
Keywords: NAFLD; bile acids; gut microbiome; gut–liver axis; incretin hormones; metabolic inflammation; short-chain fatty acids; type 2 diabetes.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures




Similar articles
-
Prescription of Controlled Substances: Benefits and Risks.2025 Jul 6. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan–. 2025 Jul 6. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan–. PMID: 30726003 Free Books & Documents.
-
Gut Microbiota-Derived Metabolites Orchestrate Metabolic Reprogramming in Diabetic Cardiomyopathy: Mechanisms and Therapeutic Frontiers.FASEB J. 2025 Sep 15;39(17):e71004. doi: 10.1096/fj.202501579RR. FASEB J. 2025. PMID: 40899744 Free PMC article. Review.
-
Synbiotics, prebiotics and probiotics for solid organ transplant recipients.Cochrane Database Syst Rev. 2022 Sep 20;9(9):CD014804. doi: 10.1002/14651858.CD014804.pub2. Cochrane Database Syst Rev. 2022. PMID: 36126902 Free PMC article.
-
The Duodenum-Centered Neurohormonal Hypothesis of Type 2 Diabetes: A Mechanistic Review and Therapeutic Perspective.Curr Issues Mol Biol. 2025 Aug 14;47(8):657. doi: 10.3390/cimb47080657. Curr Issues Mol Biol. 2025. PMID: 40864811 Free PMC article. Review.
-
Multi-strain probiotics attenuate carbohydrate-lipid metabolic dysregulation in type 2 diabetic rats via gut-liver axis modulation.mSystems. 2025 Jul 22;10(7):e0036925. doi: 10.1128/msystems.00369-25. Epub 2025 Jun 10. mSystems. 2025. PMID: 40492727 Free PMC article.
References
-
- Ong K.L., Stafford L.K., McLaughlin S.A., Boyko E.J., Vollset S.E., Smith A.E., Dalton B.E., Duprey J., Cruz J.A., Hagins H., et al. Global, Regional, and National Burden of Diabetes from 1990 to 2021, with Projections of Prevalence to 2050: A Systematic Analysis for the Global Burden of Disease Study 2021. Lancet. 2023;402:203–234. doi: 10.1016/S0140-6736(23)01301-6. - DOI - PMC - PubMed
-
- Dludla P.V., Mabhida S.E., Ziqubu K., Nkambule B.B., Mazibuko-Mbeje S.E., Hanser S., Basson A.K., Pheiffer C., Kengne A.P. Pancreatic β-Cell Dysfunction in Type 2 Diabetes: Implications of Inflammation and Oxidative Stress. World J. Diabetes. 2023;14:130–146. doi: 10.4239/wjd.v14.i3.130. - DOI - PMC - PubMed
-
- Badkas A., Pacheco M.P., Sauter T. Network Modeling Approaches for Metabolic Diseases and Diabetes. Curr. Opin. Syst. Biol. 2024;39:100530. doi: 10.1016/j.coisb.2024.100530. - DOI
-
- Phatak S.R., Saboo B., Dwivedi S., Zinzuwadia P., Panchal D., Ganguli A., Hasnani D. Sweetening Sixteen: Beyond the Ominous Octet. J. Diabetol. 2021;12:1–9. doi: 10.4103/jod.jod_9_20. - DOI
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
