

Damage-associated molecular patterns (DAMPs) in diseases: implications for therapy
- PMID: 40877572
- PMCID: PMC12394712
- DOI: 10.1186/s43556-025-00305-3
Damage-associated molecular patterns (DAMPs) in diseases: implications for therapy
Abstract
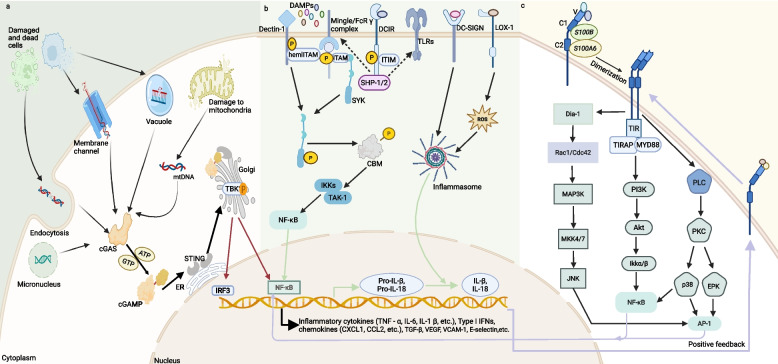
Damage-associated molecular patterns (DAMPs) are endogenous danger signal molecules released by damaged, stressed or dead cells that bind to pattern recognition receptors (PRRs), activating immune responses and inflammatory signaling pathways to play critical regulatory roles in various pathophysiological processes. This review classifies DAMPs into three major categories (protein-based, nucleic acid-based and mitochondria-derived) based on distinct molecular characteristics and biological functions, analyzing their structural features and functional differences. We systematically summarize current understanding of DAMP molecular transformation mechanisms, release pathways and recognition processes, with in-depth discussion of their pathological roles in major diseases including cancer, cardiovascular diseases and respiratory disorders. Particular emphasis is placed on the molecular recognition mechanisms between DAMPs and PRRs (TLRs, NLRs, CLRs and RAGE), and the disease regulatory networks formed by activated key signaling pathways (NF-κB, MAPK, inflammasomes and cGAS-STING). Current DAMP/PRR-targeted therapeutic strategies are comprehensively reviewed, including: modulating cell death pathways to reduce DAMP release, neutralizing DAMP activity using monoclonal antibodies, developing small-molecule inhibitors to block signaling pathways, and employing enzymatic degradation or gene silencing technologies for precise intervention. While showing promise in inflammatory and cancer disease models, these approaches face clinical translation challenges including DAMP molecular heterogeneity, inefficient drug delivery systems, and the complexity of multi-target synergistic mechanisms. Potential solutions involving nanoparticle delivery systems, AI-driven personalized treatment optimization and gene editing technologies are discussed. This review aims to provide references for developing novel therapeutics targeting the DAMP/PRR signaling axis, potentially opening new treatment avenues for cancer, neurodegenerative diseases, cardiovascular diseases and inflammatory disorders.
Keywords: Damage-associated molecular patterns; Immune response; Pathways; Pattern recognition receptors; Therapeutic strategies.
© 2025. The Author(s).
Conflict of interest statement
Declarations. Ethics approval and consent to participate: Not applicable. This scoping review did not involve human participants or animal subjects. Competing interests: The authors have no relevant financial or non-financial interests to disclose.
Figures




References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials