

A spatial long-read approach at near-single-cell resolution reveals developmental regulation of splicing and polyadenylation sites in distinct cortical layers and cell types
- PMID: 40883294
- PMCID: PMC12397408
- DOI: 10.1038/s41467-025-63301-9
A spatial long-read approach at near-single-cell resolution reveals developmental regulation of splicing and polyadenylation sites in distinct cortical layers and cell types
Abstract
Genome-wide spatial long-read approaches often lack single-cell resolution and yield limited read lengths. Here, we introduce spatial ISOform sequencing (Spl-ISO-Seq), which reveals exons and polyadenylation sites with near-single-cell resolution. Spl-ISO-Seq selects long cDNAs and doubles to triples read lengths compared to standard preparations. Adding a highly specific software tool (Spl-ISOquant) and comparing human post-mortem pre-puberty (8-11 years) to post-puberty (16-19 years) visual cortex samples, we find that cortex harbors stronger splicing and poly(A)-site regulation than white matter. However, oligodendrocyte regulation is stronger in white matter. Among cortical layers, layer 4 has the most developmentally-regulated splicing changes in excitatory neurons and in poly(A) sites. We also find repeat elements downstream of developmentally-regulated layer 4 exons. Overall, alternative splicing changes are linked to post-synaptic structure and function. These results root developmental splicing changes during puberty in specific layers and cell types. More generally, our technologies enable exciting observations for any complex tissue.
© 2025. The Author(s).
Conflict of interest statement
Competing interests: H.U.T. has presented at user meetings of 10× Genomics, Oxford Nanopore Technologies, and Pacific Biosciences, which in some cases included payment for travel and accommodations. H.U.T. has recently agreed to consult for ISOgenix Ltd., for work unrelated to the present manuscript. The other authors declare no competing interests.
Figures
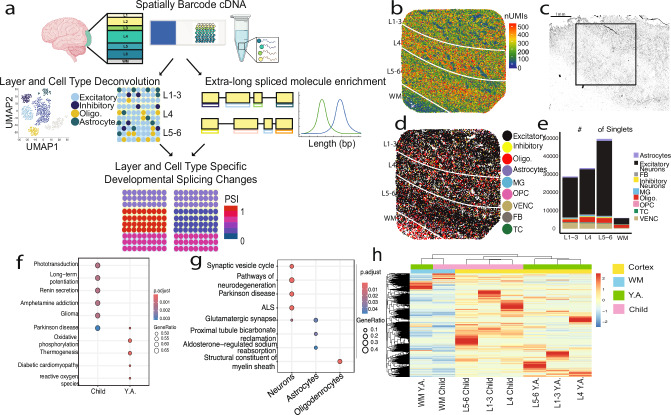


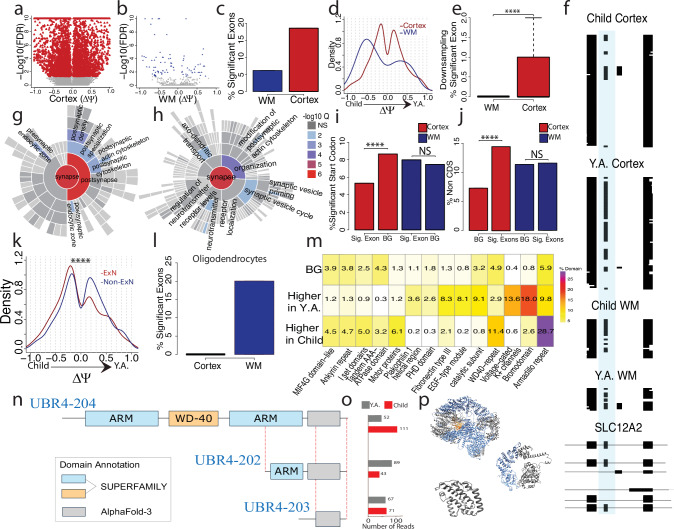


Update of
-
A spatial long-read approach at near-single-cell resolution reveals developmental regulation of splicing and polyadenylation sites in distinct cortical layers and cell types.bioRxiv [Preprint]. 2025 Jun 10:2025.06.10.658877. doi: 10.1101/2025.06.10.658877. bioRxiv. 2025. Update in: Nat Commun. 2025 Aug 29;16(1):8093. doi: 10.1038/s41467-025-63301-9. PMID: 40661641 Free PMC article. Updated. Preprint.
References
-
- Johnson, J. M. et al. Genome-wide survey of human alternative pre-mRNA splicing with exon junction microarrays. Science302, 2141–2144 (2003). - PubMed
-
- Pan, Q., Shai, O., Lee, L. J., Frey, B. J. & Blencowe, B. J. Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat. Genet.40, 1413–1415 (2008). - PubMed
MeSH terms
Grants and funding
- MIRA R35 GM152101-01/U.S. Department of Health & Human Services | NIH | National Institute of General Medical Sciences (NIGMS)
- R01 GM135247/GM/NIGMS NIH HHS/United States
- R35 GM152101/GM/NIGMS NIH HHS/United States
- 851093, SAFEBIO/EC | Horizon 2020 Framework Programme (EU Framework Programme for Research and Innovation H2020)
- U01 DA053625-01/U.S. Department of Health & Human Services | NIH | National Institute on Drug Abuse (NIDA)
- U01 DA053625/DA/NIDA NIH HHS/United States
- 2T32DA039080/U.S. Department of Health & Human Services | NIH | National Institute on Drug Abuse (NIDA)
- R01 HD111089/HD/NICHD NIH HHS/United States
- T32 DA039080/DA/NIDA NIH HHS/United States
- R01HD111089/U.S. Department of Health & Human Services | National Institutes of Health (NIH)
- RF1 MH121267/MH/NIMH NIH HHS/United States
- 1R01LM014017-01/U.S. Department of Health & Human Services | NIH | U.S. National Library of Medicine (NLM)
- R35 GM138152/GM/NIGMS NIH HHS/United States
- No. 851093, SAFEBIO/EC | Horizon 2020 Framework Programme (EU Framework Programme for Research and Innovation H2020)
- GRFP # 2139291/National Science Foundation (NSF)
- 1R01GM135247-01/U.S. Department of Health & Human Services | NIH | National Institute of General Medical Sciences (NIGMS)
- R01 LM014017/LM/NLM NIH HHS/United States
LinkOut - more resources
Full Text Sources
