

A novel splice-altering frameshift variant in the COL1A1 gene underlies osteogenesis imperfecta type I: molecular characterization of a four-generation Chinese pedigree and literature review
- PMID: 40887625
- PMCID: PMC12398993
- DOI: 10.1186/s40246-025-00816-8
A novel splice-altering frameshift variant in the COL1A1 gene underlies osteogenesis imperfecta type I: molecular characterization of a four-generation Chinese pedigree and literature review
Abstract
Backgroud: Osteogenesis imperfecta (OI) is a phenotypically and genetically heterogeneous group of inherited connective tissue disorder. This investigation aims to elucidate the molecular etiology underlying a four-generation Chinese family affected by OI.
Methods: Whole-exome sequencing was employed to identify pathogenic variants in the proband, with subsequent Sanger sequencing performed for familial co-segregation analysis. A minigene assay was conducted to investigate the effect of variant on splicing patterns. The pathogenic potential of variant was evaluated through protein structural modeling and HEK293 cell-based functional studies. COL1A1 splicing variants were further collated to analyze its occurrence frequency across geographically diverse OI cohorts, intronic distribution patterns and potential hotpots for mild versus severe subtypes.
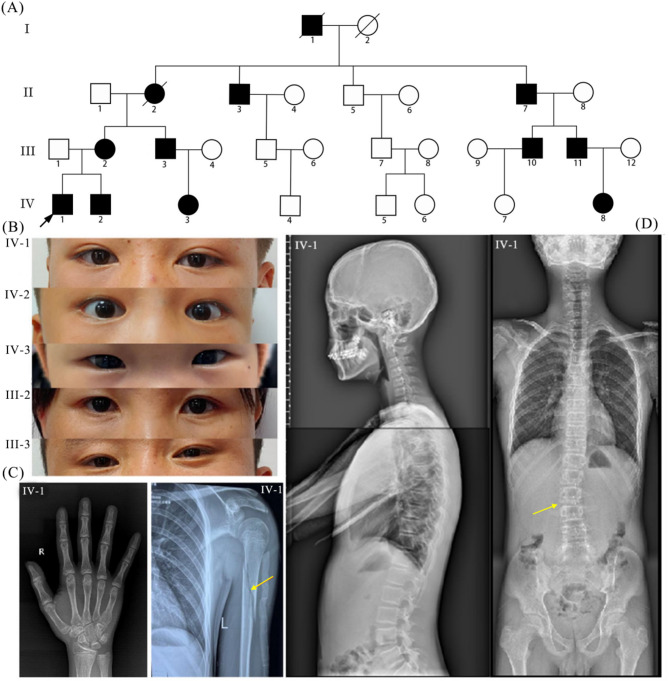
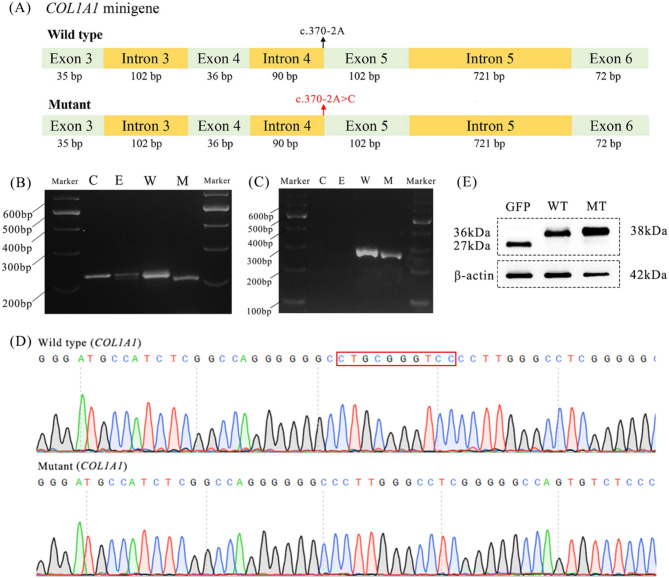
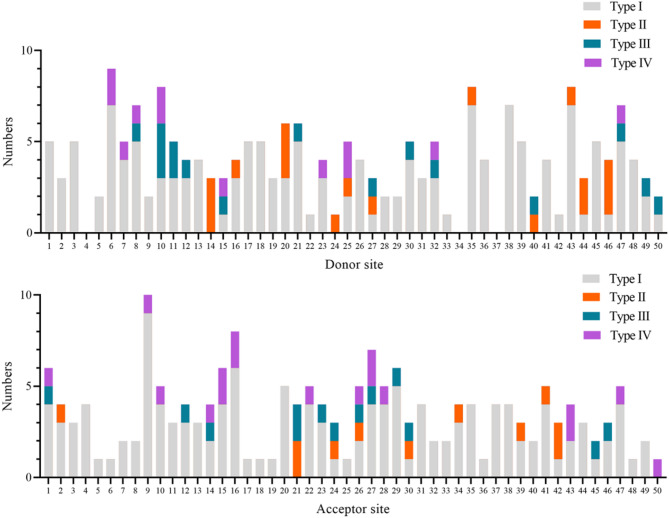
Results: Multiple affected members within an non-consanguineous Chinese pedigree exhibited clinical manifestations fitting OI-associated phenotypic spectrum. A novel heterozygous COL1A1 splicing variant (c.370-2A > C) inherited from the mother was identified in the proband. The splicing variant altered the canonical acceptor site (AG) at the intron 4-exon 5 junction, activating a adjacent cryptic splicing site in exon 5. This abberrant splicing event introduced a frameshift variant (c.370_379delGGACCCGCAG), and generated a premature termination codon that truncates the COL1A1 protein (p.Gly124Alafs*138). AlphaFold3-based protein structural modeling revealed the loss of the triple-helical domain in this truncated protein. In vitro functional assays showed that mRNA and protein expression levels of mutant COL1A1 were significantly increased than wild-type COL1A1 (p < 0.05). Comprehensive literature analysis indicated that COL1A1 splicing variants account for < 10% of variants in OI cohorts from the vast majority of regions. The acceptor site of intron 9 and the donor sites of intron 35 are hotspots for COL1A1 splicing variant occurrence. Moreover, the majority of COL1A1 splicing variants, expecially those proximal to the 5' and 3' terminal regions, result in mild manifestations of OI type I, whereas variants at donor sites of introns 14, 20, and 46, may be candidate hotspots for lethal OI type II.
Conclusions: Our study revealed the pathogenic mechanism of a novel COL1A1 splicing variant in a four-generation Chinese family with OI, and provided updated data on COL1A1 splicing variants and its potential hotpots for mild versus severe OI subtypes.
Keywords: COL1A1; Minigene assay; Osteogenesis imperfecta; Splicing variant; Whole-exome sequencing.
© 2025. The Author(s).
Conflict of interest statement
Declarations. Ethics approval and consent to participate: The research protocol was reviewed and ethically approved by the Institutional Review Board of the Affiliated Hospital of Jining Medical University (Approval No. 2019C003), in full compliance with the ethical principles established in the Declaration of Helsinki. Written informed consent was obtained from all participants prior to the collection of clinical data and publication of research findings. Consent for publication: Written informed consent, including the permission to publish facial photos, has been obtained from the individual persons or their legal guardian. Competing interests: The authors declare no competing interests.
Figures



Similar articles
-
COL1A1- and COL1A2-Related Osteogenesis Imperfecta.2005 Jan 28 [updated 2025 May 29]. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993–2025. 2005 Jan 28 [updated 2025 May 29]. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993–2025. PMID: 20301472 Free Books & Documents. Review.
-
A novel splicing pathogenic variant in COL1A1 causing osteogenesis imperfecta (OI) type I in a Chinese family.Mol Genet Genomic Med. 2020 Sep;8(9):e1366. doi: 10.1002/mgg3.1366. Epub 2020 Jun 25. Mol Genet Genomic Med. 2020. PMID: 32588564 Free PMC article.
-
Anti-transforming growth factor-β treatment shows increased bone mass and strength in a novel mouse model for osteogenesis imperfecta type I.J Bone Miner Res. 2025 Jun 25;40(7):881-890. doi: 10.1093/jbmr/zjaf068. J Bone Miner Res. 2025. PMID: 40358008
-
A new Col1a1 conditional knock-in mouse model to study osteogenesis imperfecta.J Bone Miner Res. 2024 Dec 31;40(1):114-124. doi: 10.1093/jbmr/zjae189. J Bone Miner Res. 2024. PMID: 39566076
-
[Clinical phenotype and genotypic analysis of a four-generation Chinese pedigree affected with Stickler syndrome and a literature review].Zhonghua Yi Xue Yi Chuan Xue Za Zhi. 2025 Jun 10;42(6):684-690. doi: 10.3760/cma.j.cn511374-20250418-00235. Zhonghua Yi Xue Yi Chuan Xue Za Zhi. 2025. PMID: 40763965 Review. Chinese.
References
-
- Wei SS, Yao YY, Shu M, Gao L, Zhao JJ, Li TY, et al. Genotype-phenotype relationship and follow-up analysis of a Chinese cohort with osteogenesis imperfecta. Endocr Pract. 2022;28(8):760–6. - PubMed
-
- Marini JC, Forlino A, Bächinger HP, Bishop NJ, Byers PH, De Paepe A, et al. Osteogenesis imperfecta. Nat Rev Dis Primers. 2017;3:17052. - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
