

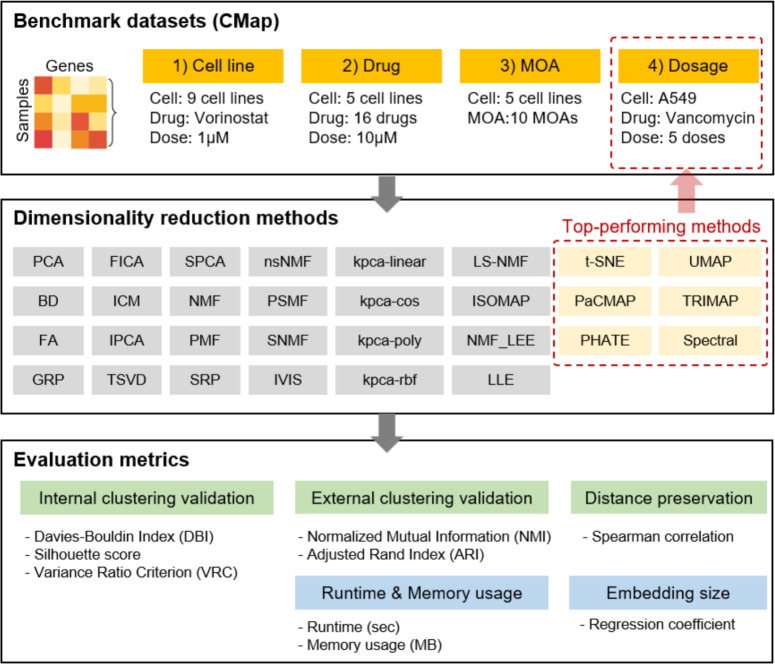
Benchmarking of dimensionality reduction methods to capture drug response in transcriptome data
- PMID: 40890192
- PMCID: PMC12402208
- DOI: 10.1038/s41598-025-12021-7
Benchmarking of dimensionality reduction methods to capture drug response in transcriptome data
Abstract
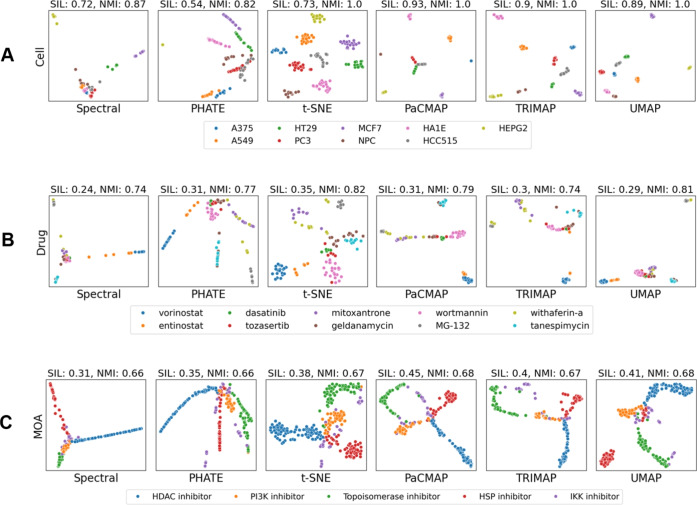
Drug-induced transcriptomic data are crucial for understanding molecular mechanisms of action (MOAs), predicting drug efficacy, and identifying off-target effects. However, their high dimensionality presents challenges for analysis and interpretation. Dimensionality reduction (DR) methods simplify such data, enabling efficient analysis and visualization. Despite their importance, few studies have evaluated the performance of DR methods specifically for drug-induced transcriptomic data. We tested the DR methods across four distinct experimental conditions using data from the Connectivity Map (CMap) dataset, which includes different cell lines, drugs, MOA, and drug dosages. t-distributed Stochastic Neighbor Embedding (t-SNE), Uniform Manifold Approximation and Projection (UMAP), Pairwise Controlled Manifold Approximation (PaCMAP), and TRIMAP outperformed other methods in preserving both local and global biological structures, particularly in separating distinct drug responses and grouping drugs with similar molecular targets. However, most methods struggled with detecting subtle dose-dependent transcriptomic changes, where Spectral, Potential of Heat-diffusion for Affinity-based Trajectory Embedding (PHATE), and t-SNE showed stronger performance. Standard parameter settings limited the optimal performance of DR methods, highlighting the need for further exploration of hyperparameter optimization. Our study provides valuable insights into the strengths and limitations of various DR methods for analyzing drug-induced transcriptomic data. While t-SNE, UMAP, and PaCMAP are well-suited for studying discrete drug responses, further refinement is needed for detecting subtle dose-dependent changes. This study highlights the importance of selecting the DR method to accurately analyze drug-induced transcriptomic data.
Keywords: CMap data; Dimension reduction; Drug-induced transcriptome; RNA-seq analysis.
© 2025. The Author(s).
Conflict of interest statement
Declarations. Competing interests: The authors declare no competing interests.
Figures





References
-
- Kwon, O.-S., Kim, W., Cha, H.-J. & Lee, H. In silico drug repositioning: From large-scale transcriptome data to therapeutics. Arch. Pharmacal Res.42, 879–889 (2019). - PubMed
-
- Yang, Y. et al. Dimensionality reduction by umap reinforces sample heterogeneity analysis in bulk transcriptomic data. Cell Rep.36 (2021). - PubMed
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
