

ATF6 activation alters colonic lipid metabolism causing tumour-associated microbial adaptation
- PMID: 40890536
- PMCID: PMC12460170
- DOI: 10.1038/s42255-025-01350-6
ATF6 activation alters colonic lipid metabolism causing tumour-associated microbial adaptation
Abstract
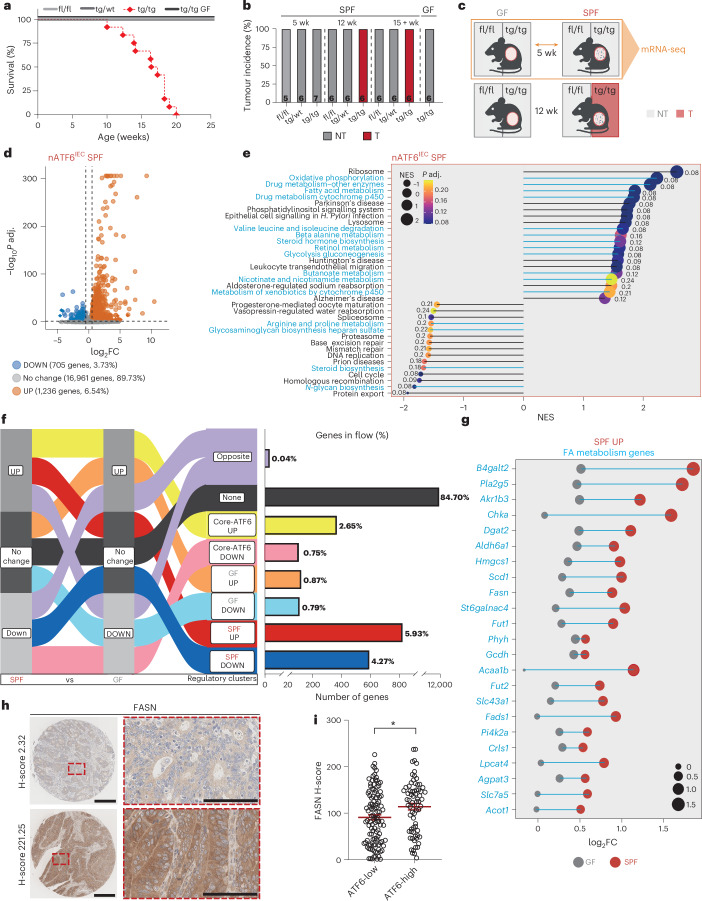
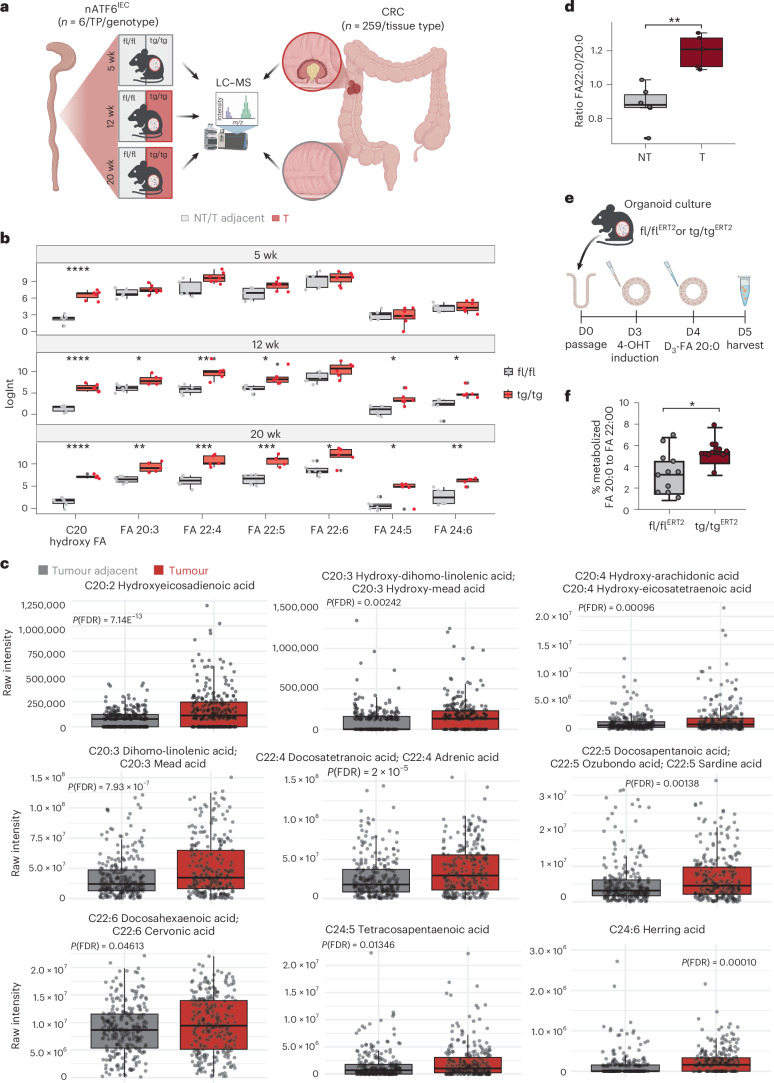
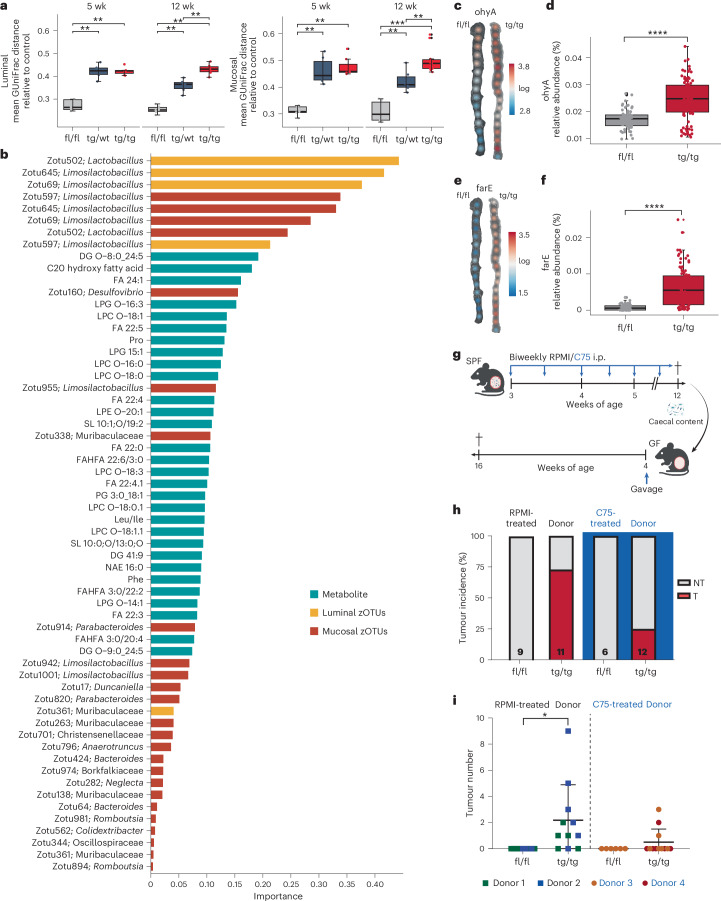
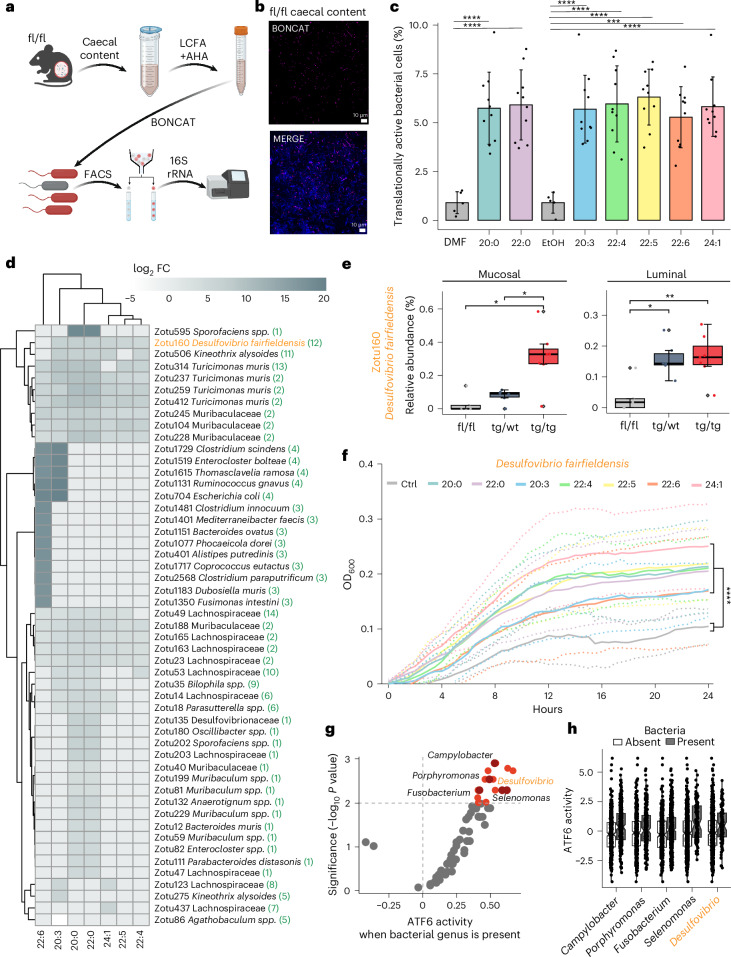
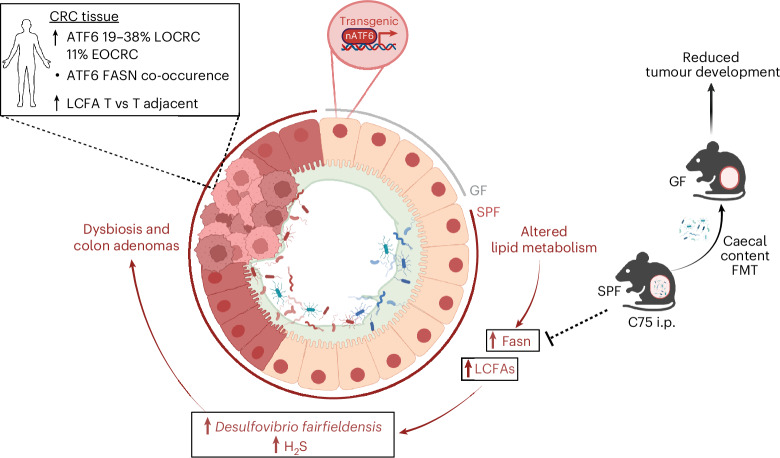
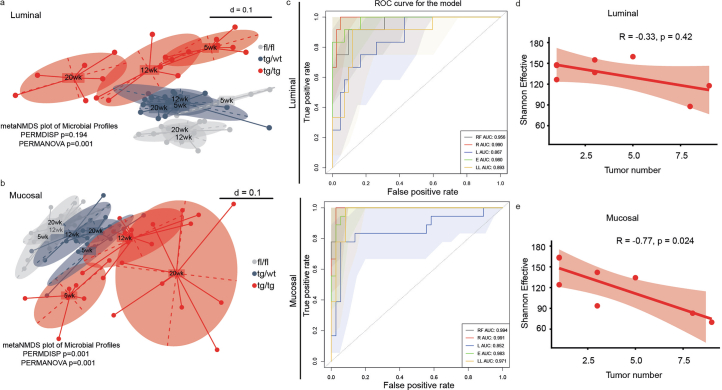
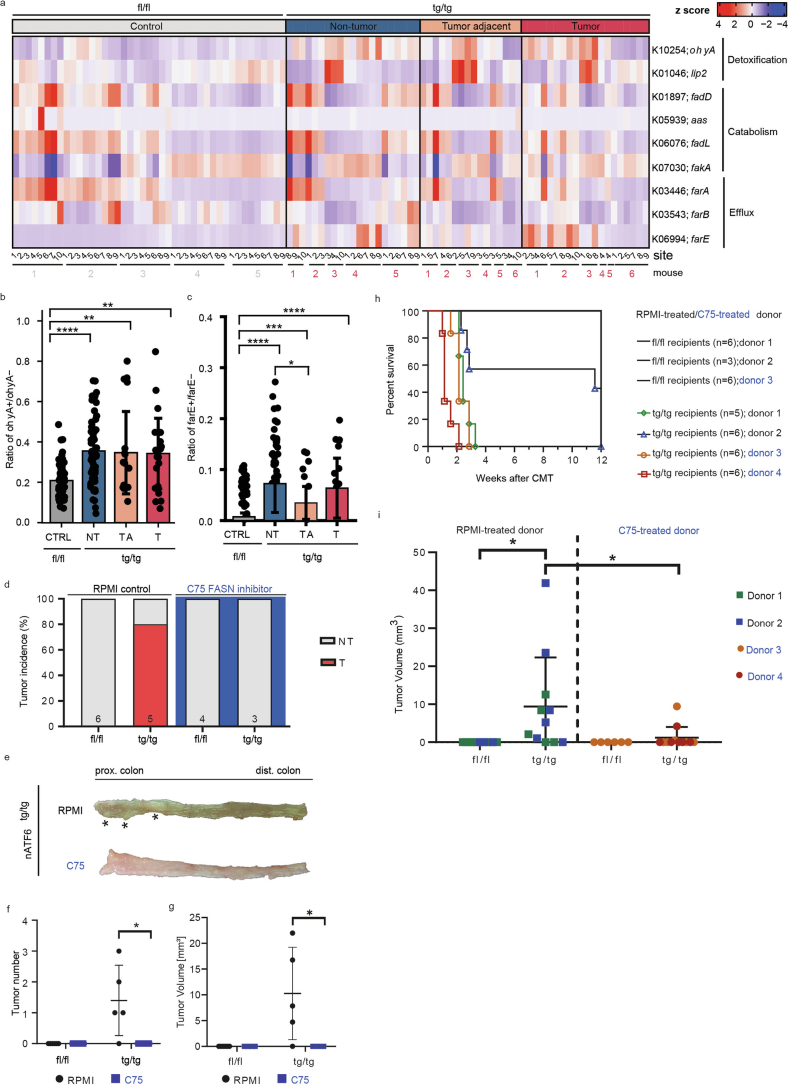
Endoplasmic reticulum unfolded protein responses contribute to cancer development, with activating transcription factor 6 (ATF6) involved in microbiota-dependent tumorigenesis. Here we show the clinical relevance of ATF6 in individuals with early-onset and late colorectal cancer, and link ATF6 signalling to changes in lipid metabolism and intestinal microbiota. Transcriptional analysis in intestinal epithelial cells of ATF6 transgenic mice (nATF6IEC) identifies bacteria-specific changes in cellular metabolism enriched for fatty acid biosynthesis. Untargeted metabolomics and isotype labelling confirm ATF6-related enrichment of long-chain fatty acids in colonic tissue of humans, mice and organoids. FASN inhibition and microbiota transfer in germ-free nATF6IEC mice confirm the causal involvement of ATF6-induced lipid alterations in tumorigenesis. The selective expansion of tumour-relevant microbial taxa, including Desulfovibrio fairfieldensis, is mechanistically linked to long-chain fatty acid exposure using bioorthogonal non-canonical amino acid tagging, and growth analysis of Desulfovibrio isolates. We postulate chronic ATF6 signalling to select for tumour-promoting microbiota by altering lipid metabolism.
© 2025. The Author(s).
Conflict of interest statement
Competing interests: The authors declare no competing interests.
Figures









References
-
- Braakman, I. & Bulleid, N. J. Protein folding and modification in the mammalian endoplasmic reticulum. Annu. Rev. Biochem.80, 71–99 (2011). - PubMed
-
- Rath, E., Moschetta, A. & Haller, D. Mitochondrial function - gatekeeper of intestinal epithelial cell homeostasis. Nat. Rev. Gastroenterol. Hepatol.15, 497–516 (2018). - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
