

Functional phenotyping of genomic variants using joint multiomic single-cell DNA-RNA sequencing
- PMID: 40890552
- PMCID: PMC12510883
- DOI: 10.1038/s41592-025-02805-0
Functional phenotyping of genomic variants using joint multiomic single-cell DNA-RNA sequencing
Abstract
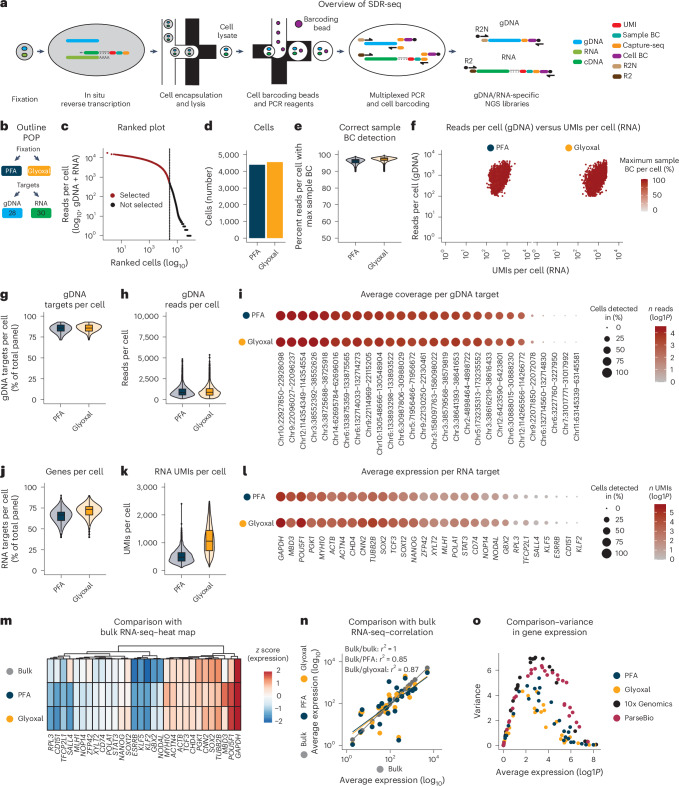
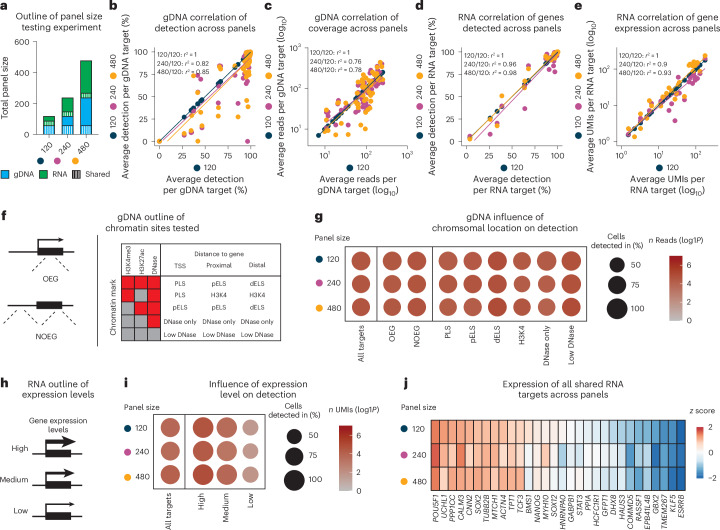
Genetic variants (both coding and noncoding) can impact gene function and expression, driving disease mechanisms such as cancer progression. The systematic study of endogenous genetic variants is hindered by inefficient precision editing tools, combined with technical limitations in confidently linking genotypes to gene expression at single-cell resolution. We developed single-cell DNA-RNA sequencing (SDR-seq) to simultaneously profile up to 480 genomic DNA loci and genes in thousands of single cells, enabling accurate determination of coding and noncoding variant zygosity alongside associated gene expression changes. Using SDR-seq, we associate coding and noncoding variants with distinct gene expression in human induced pluripotent stem cells. Furthermore, we demonstrate that in primary B cell lymphoma samples, cells with a higher mutational burden exhibit elevated B cell receptor signaling and tumorigenic gene expression. SDR-seq provides a powerful platform to dissect regulatory mechanisms encoded by genetic variants, advancing our understanding of gene expression regulation and its implications for disease.
© 2025. The Author(s).
Conflict of interest statement
Competing interests: L.M.S. is cofounder and shareholder of Sophia Genetics and founder and board member of LevitasBio and Recombia Biosciences. L.M.S. and D.L. have submitted a patent application on ‘High-throughput multiomic readout of RNA and genomic DNA within single cells’ (PCT/US2024/029950). D.F. is a cofounder and board member of Anthropocene Bio. The remaining authors declare no competing interests.
Figures











References
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
