

Gene expression variation across genetically identical individuals predicts reproductive traits
- PMID: 40891666
- PMCID: PMC12404612
- DOI: 10.7554/eLife.106525
Gene expression variation across genetically identical individuals predicts reproductive traits
Abstract
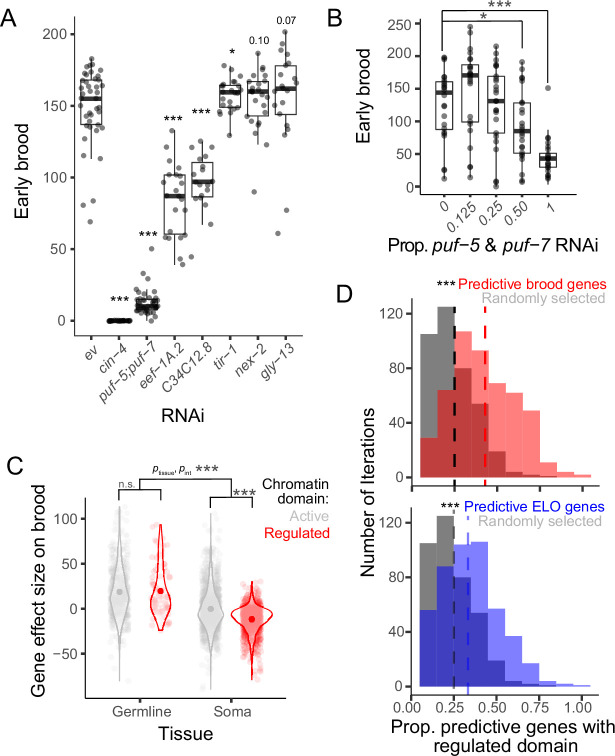
In recent decades, genome-wide association studies (GWAS) have been the major approach to understand the biological basis of individual differences in traits and diseases. However, GWAS approaches have limited predictive power to explain individual differences, particularly for complex traits and diseases in which environmental factors play a substantial role in their etiology. Indeed, individual differences persist even in genetically identical individuals, although fully separating genetic and environmental causation is difficult in most organisms. To understand the basis of individual differences in the absence of genetic differences, we measured two quantitative reproductive traits in 180 genetically identical young adult Caenorhabditis elegans roundworms in a shared environment and performed single-individual transcriptomics on each worm. We identified hundreds of genes for which expression variation was strongly associated with reproductive traits, some of which depended on individuals' historical environments and some of which was random. Multiple small sets of genes together were highly predictive of reproductive traits, explaining on average over half and over a quarter of variation in the two traits. We manipulated mRNA levels of predictive genes to identify a set of causal genes, demonstrating the utility of this approach for both prediction and understanding underlying biology. Finally, we found that the chromatin environment of predictive genes was enriched for H3K27 trimethylation, suggesting that gene expression variation may be driven in part by chromatin structure. Together, this work shows that individual, non-genetic differences in gene expression are both highly predictive and causal in shaping reproductive traits.
Keywords: C. elegans; environmental variation; epigenetics; gene expression; genetics; genomics; reproduction.
© 2025, Webster et al.
Conflict of interest statement
AW, JW, EJ, PS, PP No competing interests declared
Figures






Update of
-
Gene expression variation across genetically identical individuals predicts reproductive traits.bioRxiv [Preprint]. 2025 Jun 6:2023.10.13.562270. doi: 10.1101/2023.10.13.562270. bioRxiv. 2025. Update in: Elife. 2025 Sep 02;14:RP106525. doi: 10.7554/eLife.106525. PMID: 37873136 Free PMC article. Updated. Preprint.
References
-
- Ahringer J. Reverse Genetics. WormBook; 2006. - DOI
-
- Alsheikh AJ, Wollenhaupt S, King EA, Reeb J, Ghosh S, Stolzenburg LR, Tamim S, Lazar J, Davis JW, Jacob HJ. The landscape of GWAS validation; systematic review identifying 309 validated non-coding variants across 130 human diseases. BMC Medical Genomics. 2022;15:74. doi: 10.1186/s12920-022-01216-w. - DOI - PMC - PubMed
-
- Barban N, Jansen R, de Vlaming R, Vaez A, Mandemakers JJ, Tropf FC, Shen X, Wilson JF, Chasman DI, Nolte IM, Tragante V, van der Laan SW, Perry JRB, Kong A, Ahluwalia TS, Albrecht E, Yerges-Armstrong L, Atzmon G, Auro K, Ayers K, Bakshi A, Ben-Avraham D, Berger K, Bergman A, Bertram L, Bielak LF, Bjornsdottir G, Bonder MJ, Broer L, Bui M, Barbieri C, Cavadino A, Chavarro JE, Turman C, Concas MP, Cordell HJ, Davies G, Eibich P, Eriksson N, Esko T, Eriksson J, Falahi F, Felix JF, Fontana MA, Franke L, Gandin I, Gaskins AJ, Gieger C, Gunderson EP, Guo X, Hayward C, He C, Hofer E, Huang H, Joshi PK, Kanoni S, Karlsson R, Kiechl S, Kifley A, Kluttig A, Kraft P, Lagou V, Lecoeur C, Lahti J, Li-Gao R, Lind PA, Liu T, Makalic E, Mamasoula C, Matteson L, Mbarek H, McArdle PF, McMahon G, Meddens SFW, Mihailov E, Miller M, Missmer SA, Monnereau C, van der Most PJ, Myhre R, Nalls MA, Nutile T, Kalafati IP, Porcu E, Prokopenko I, Rajan KB, Rich-Edwards J, Rietveld CA, Robino A, Rose LM, Rueedi R, Ryan KA, Saba Y, Schmidt D, Smith JA, Stolk L, Streeten E, Tönjes A, Thorleifsson G, Ulivi S, Wedenoja J, Wellmann J, Willeit P, Yao J, Yengo L, Zhao JH, Zhao W, Zhernakova DV, Amin N, Andrews H, Balkau B, Barzilai N, Bergmann S, Biino G, Bisgaard H, Bønnelykke K, Boomsma DI, Buring JE, Campbell H, Cappellani S, Ciullo M, Cox SR, Cucca F, Toniolo D, Davey-Smith G, Deary IJ, Dedoussis G, Deloukas P, van Duijn CM, de Geus EJC, Eriksson JG, Evans DA, Faul JD, Sala CF, Froguel P, Gasparini P, Girotto G, Grabe HJ, Greiser KH, Groenen PJF, de Haan HG, Haerting J, Harris TB, Heath AC, Heikkilä K, Hofman A, Homuth G, Holliday EG, Hopper J, Hyppönen E, Jacobsson B, Jaddoe VWV, Johannesson M, Jugessur A, Kähönen M, Kajantie E, Kardia SLR, Keavney B, Kolcic I, Koponen P, Kovacs P, Kronenberg F, Kutalik Z, La Bianca M, Lachance G, Iacono WG, Lai S, Lehtimäki T, Liewald DC, Lindgren CM, Liu Y, Luben R, Lucht M, Luoto R, Magnus P, Magnusson PKE, Martin NG, McGue M, McQuillan R, Medland SE, Meisinger C, Mellström D, Metspalu A, Traglia M, Milani L, Mitchell P, Montgomery GW, Mook-Kanamori D, de Mutsert R, Nohr EA, Ohlsson C, Olsen J, Ong KK, Paternoster L, Pattie A, Penninx B, Perola M, Peyser PA, Pirastu M, Polasek O, Power C, Kaprio J, Raffel LJ, Räikkönen K, Raitakari O, Ridker PM, Ring SM, Roll K, Rudan I, Ruggiero D, Rujescu D, Salomaa V, Schlessinger D, Schmidt H, Schmidt R, Schupf N, Smit J, Sorice R, Spector TD, Starr JM, Stöckl D, Strauch K, Stumvoll M, Swertz MA, Thorsteinsdottir U, Thurik AR, Timpson NJ, Tung JY, Uitterlinden AG, Vaccargiu S, Viikari J, Vitart V, Völzke H, Vollenweider P, Vuckovic D, Waage J, Wagner GG, Wang JJ, Wareham NJ, Weir DR, Willemsen G, Willeit J, Wright AF, Zondervan KT, Stefansson K, Krueger RF, Lee JJ, Benjamin DJ, Cesarini D, Koellinger PD, den Hoed M, Snieder H, Mills MC, BIOS Consortium. LifeLines Cohort Study Genome-wide analysis identifies 12 loci influencing human reproductive behavior. Nature Genetics. 2016;48:1462–1472. doi: 10.1038/ng.3698. - DOI - PMC - PubMed
-
- Bates D, Mächler M, Bolker B, Walker S. Fitting linear mixed-effects models using lme4. Journal of Statistical Software. 2015;67:1–48. doi: 10.18637/jss.v067.i01. - DOI
MeSH terms
Associated data
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
