

This is a preprint.
A unified network systems approach uncovers a core novel program underlying T follicular helper cell differentiation
- PMID: 40894583
- PMCID: PMC12393391
- DOI: 10.1101/2025.08.19.670906
A unified network systems approach uncovers a core novel program underlying T follicular helper cell differentiation
Abstract
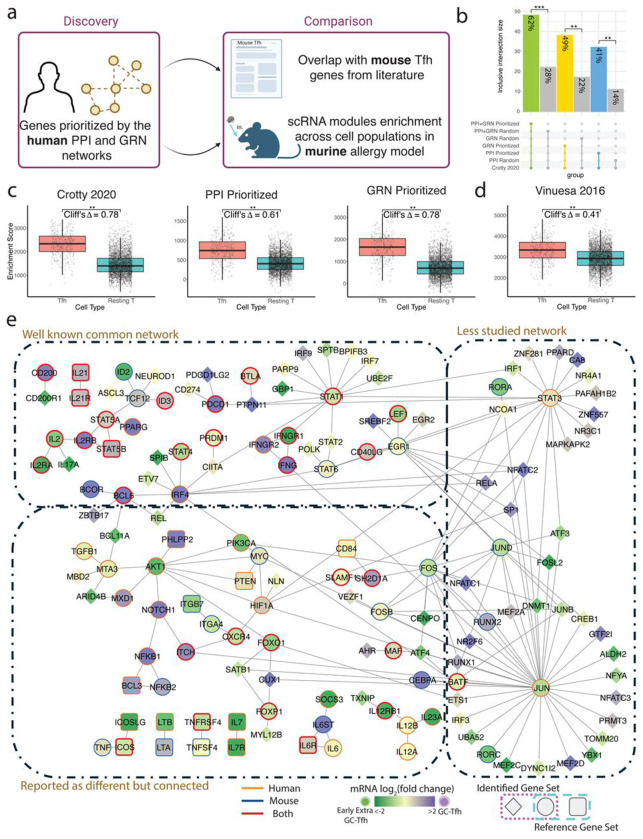
T follicular helper (Tfh) cells are central to the adaptive immune response and exhibit remarkable functional diversity and plasticity. The complex nature of Tfh cell populations, inconsistent findings across experimental systems and potential differences across species have fueled ongoing debate regarding core regulatory pathways that govern Tfh differentiation. Many studies have experimentally investigated individual proteins and circuits involved in Tfh differentiation in limited contexts, each providing only a partial understanding of the process. To address this, we adopted a novel multi-scale network systems approach that incorporates both regulatory and protein-protein interactions. Our approach integrates diverse data types, captures regulation across multiple levels of immune system organization, and recapitulates known drivers. Further, we discover a core Tfh gene set that is conserved across tissue types and disease contexts, and is consistent across data modalities - bulk, single-cell and spatial. While components of this set have been individually reported, a novel aspect of our work lies in the discovery, characterization, and connectivity of this core signature using a single unbiased approach. Using this method, we also uncover a novel function of IL-12, a molecule with reported conflicting functions, in the regulation of Tfh differentiation. Notably, we find that, in both humans and mice, IL-12 is permissive for the differentiation of Tfh precursors, but blocks subsequent differentiation into GC Tfh cells. Overall, this work elucidates novel networks with unexplored roles in governing Tfh cell differentiation across species and tissues, paving the way for novel -therapeutic interventions.
Figures










References
-
- Cosmi L., Maggi L., Santarlasci V., Liotta F. & Annunziato F. T helper cells plasticity in inflammation: T Helper Cells Plasticity in Inflammation. Cytometry A 85, 36–42 (2014). - PubMed
-
- Zhou L., Chong M. M. W. & Littman D. R. Plasticity of CD4+ T cell lineage differentiation. Immunity 30, 646–655 (2009). - PubMed
-
- Ruterbusch M., Pruner K. B., Shehata L. & Pepper M. In vivo CD4+ T cell differentiation and function: Revisiting the Th1/Th2 paradigm. Annu. Rev. Immunol. 38, 705–725 (2020). - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous