

This is a preprint.
DFFB suppresses interferon to enable cancer persister cell regrowth
- PMID: 40894800
- PMCID: PMC12393451
- DOI: 10.1101/2025.08.15.670603
DFFB suppresses interferon to enable cancer persister cell regrowth
Update in
-
DNA fragmentation factor B suppresses interferon to enable cancer persister cell regrowth.Nat Cell Biol. 2025 Dec;27(12):2143-2151. doi: 10.1038/s41556-025-01810-x. Epub 2025 Nov 17. Nat Cell Biol. 2025. PMID: 41249572 Free PMC article.
Abstract
Oncogene targeted cancer therapies can provide deep responses but frequently suffer from acquired resistance.1 Therapeutic approaches to treat tumours which have acquired drug resistance are complicated by continual tumour evolution and multiple co-occurring resistance mechanisms.2,3 Rather than treating resistance after it emerges, it may possible to prevent it by inhibiting the adaptive processes which initiate resistance but these are poorly understood.4 Here we report that residual cancer persister cells that survive oncogene targeted therapy are growth arrested by drug stress-induced intrinsic Type I interferon (IFN) signaling. To escape growth arrest, persister cells leverage apoptotic machinery to transcriptionally suppress interferon-stimulated genes (ISGs). Mechanistically, persister cells sublethally engage apoptotic caspases to activate DNA endonuclease DNA Fragmentation Factor B (DFFB, also known as Caspase-Activated DNase (CAD)) which induces DNA damage, mutagenesis, and stress response factor Activating Transcription Factor 3 (ATF3). ATF3 limits Activator Protein-1 (AP1)-mediated ISG expression sufficiently to allow persister cell regrowth. Persister cells deficient in DFFB or ATF3 exhibit high ISG expression and are consequently unable to regrow. Therefore, sublethal apoptotic stress paradoxically promotes regrowth of residual cancer cells that survive drug treatment.
Conflict of interest statement
Competing interests: M.J.H. is a cofounder, consultant, and research funding recipient of BridgeBio subsidiary Ferro Therapeutics.
Figures
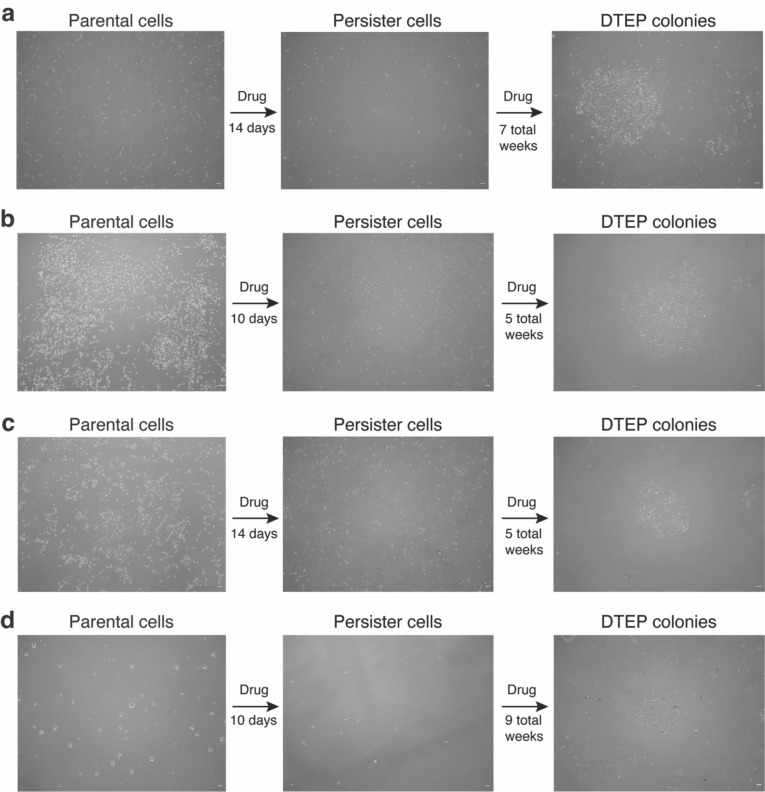




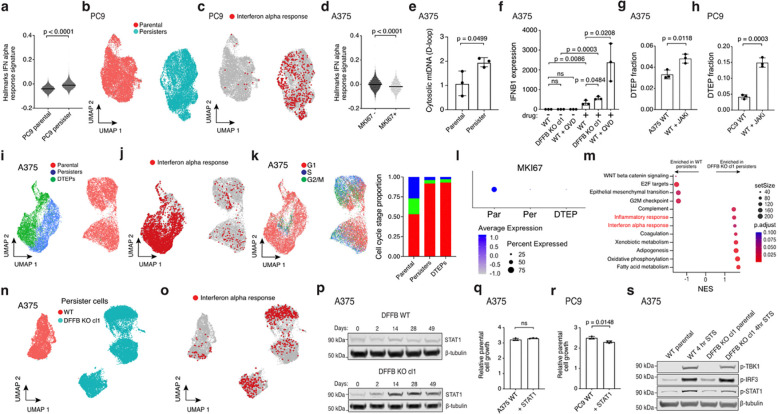
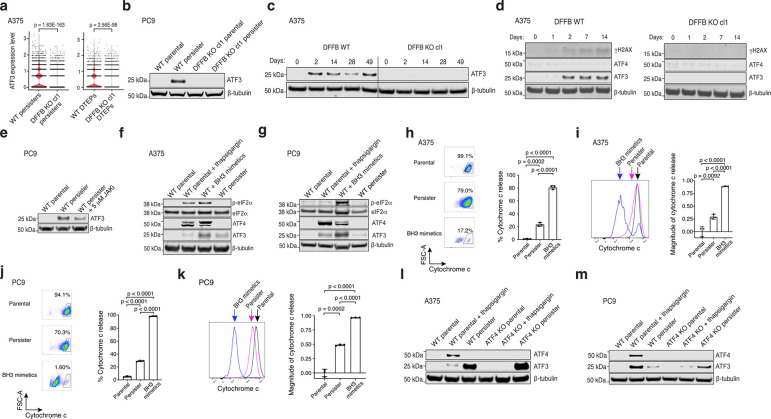





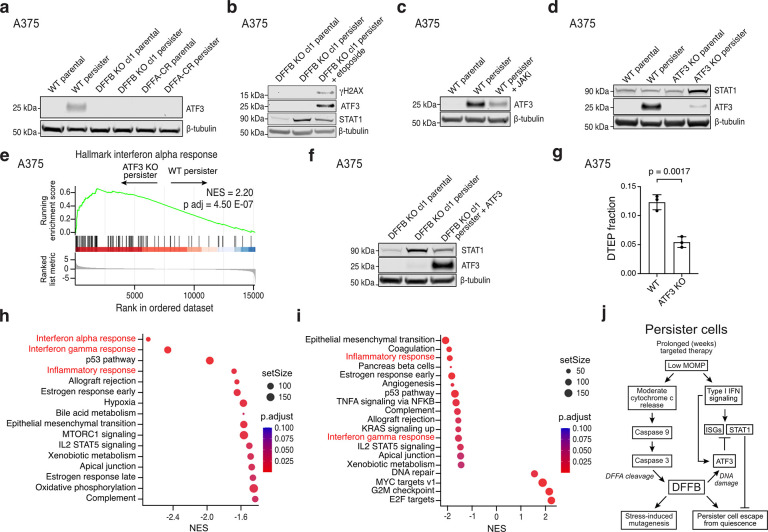
References
-
- Dagogo-Jack I. & Shaw A. T. Tumour heterogeneity and resistance to cancer therapies. Nat. Rev. Clin. Oncol. 15, 81–94 (2018). - PubMed
-
- Romano E. et al. Identification of Multiple Mechanisms of Resistance to Vemurafenib in a Patient with BRAFV600E-Mutated Cutaneous Melanoma Successfully Rechallenged after Progression. Clin. Cancer Res. 19, 5749–5757 (2013). - PubMed
-
- Mullard A. Stemming the tide of drug resistance in cancer. Nat. Rev. Drug Discov. 19, 221–223 (2020). - PubMed
Methods References:
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous