

Simultaneous STING and lymphotoxin-β receptor activation induces B cell responses in tertiary lymphoid structures to potentiate antitumor immunity
- PMID: 40897896
- PMCID: PMC12479350
- DOI: 10.1038/s41590-025-02259-8
Simultaneous STING and lymphotoxin-β receptor activation induces B cell responses in tertiary lymphoid structures to potentiate antitumor immunity
Abstract
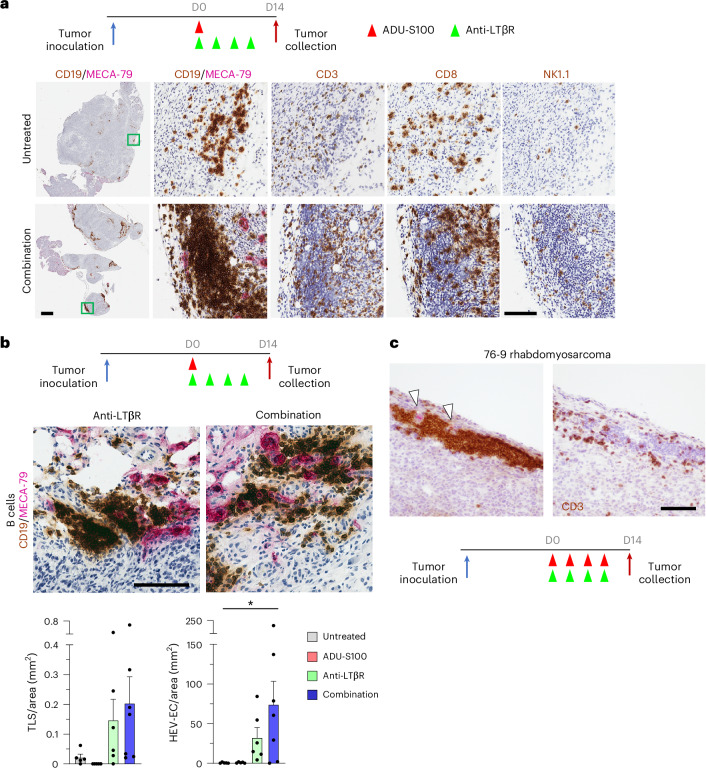
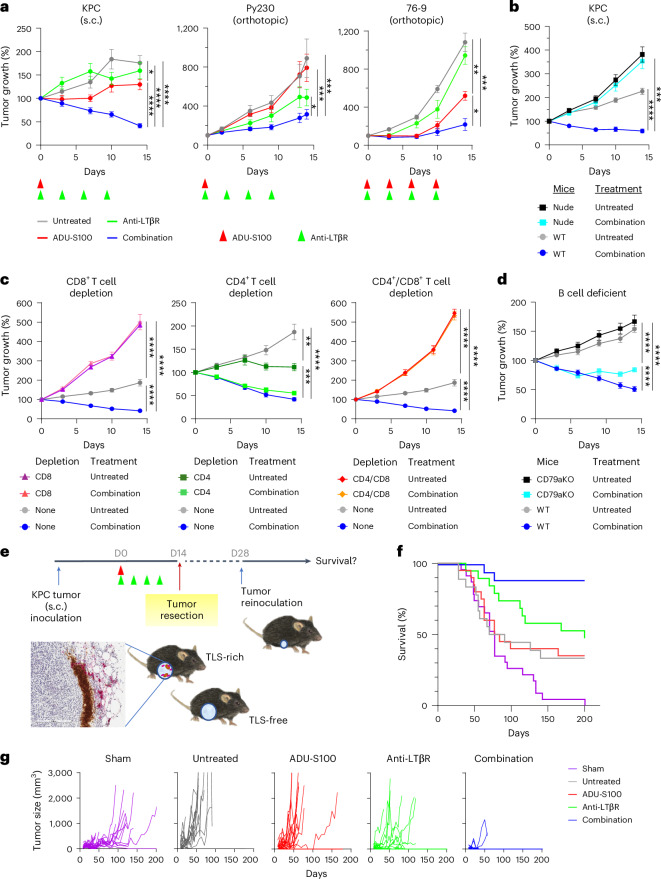
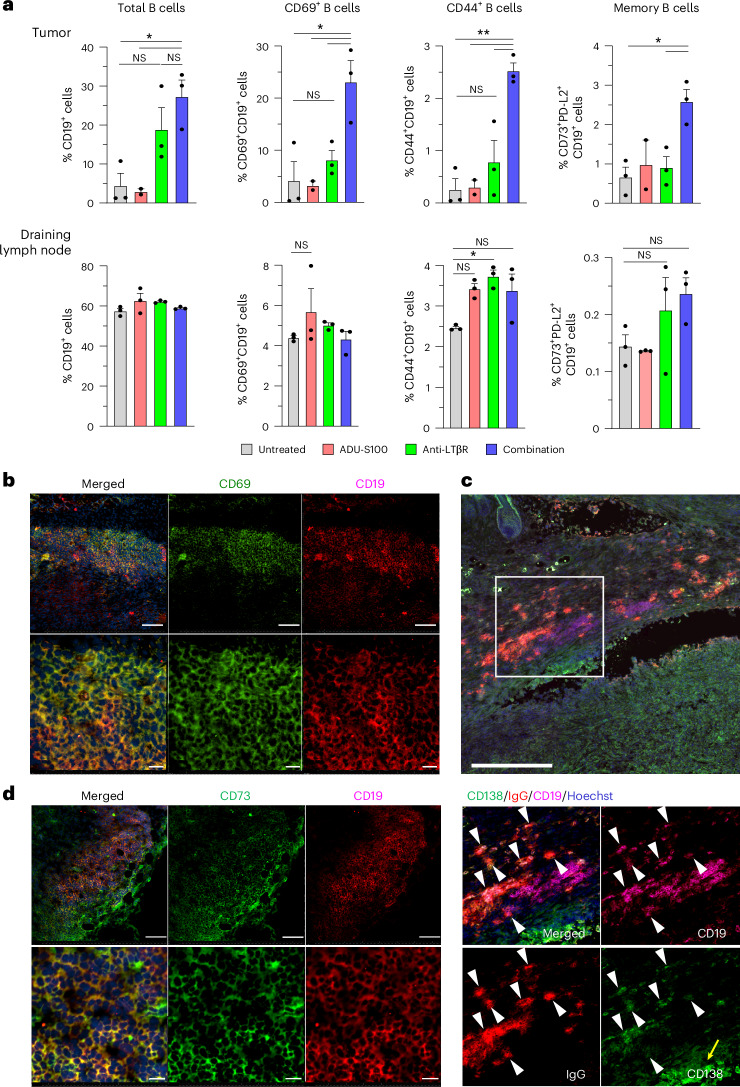
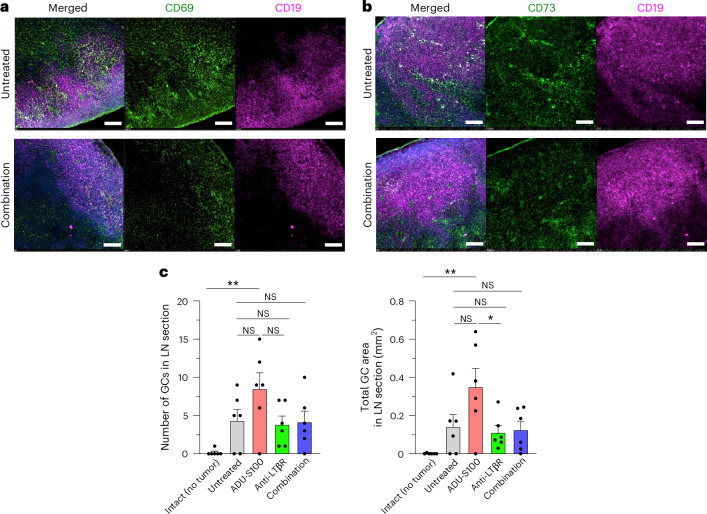
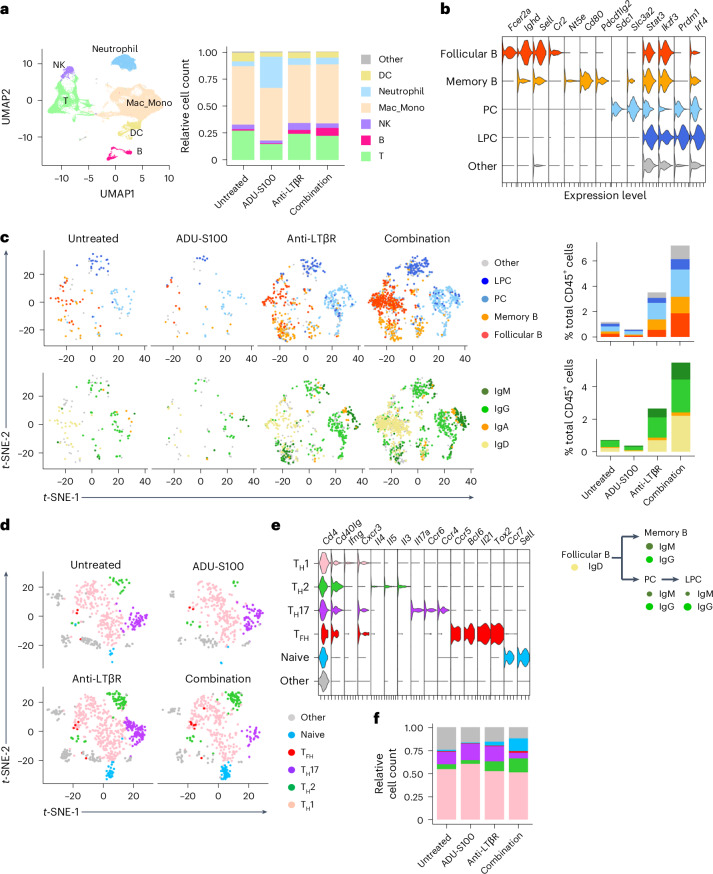
B cell-rich tertiary lymphoid structures (TLS) are associated with favorable prognosis and positive response to immunotherapy in cancer. Here we show that simultaneous activation of innate immune effectors, STING and lymphotoxin-β receptor (LTβR), results in CD8+ T cell-dependent tumor suppression while inducing high endothelial venule development and germinal center-like B cell responses in tumors to generate functional TLS in a T cell-dependent manner. In a neoadjuvant setting, activation of STING and LTβR by their agonists effectively immunized mice against tumor recurrence, leading to long-term survival. STING activation alone was insufficient for inducing B cell-containing TLS or eliciting long-term therapeutic effects. However, when combined with LTβR activation, it improved the fitness of TLS with B cell expansion and maturation to IgG-producing long-lived plasma cells and memory cells, increased CD4+ T cell recruitment and memory CD8+ T cell expansion, and shifted the TH2/TH17 balance, resulting in the potentiation of humoral and cellular immunity against tumors. These findings suggest a therapeutic approach of simultaneously activating STING and lymphotoxin pathways.
© 2025. The Author(s).
Conflict of interest statement
Competing interests: C.F.W. is a co-inventor on patents related to this research (patent no. US 8,974,787). The other authors declare no competing interests.
Figures













References
-
- Martinet, L. et al. Human solid tumors contain high endothelial venules: association with T- and B-lymphocyte infiltration and favorable prognosis in breast cancer. Cancer Res.71, 5678–5687 (2011). - PubMed
-
- Schumacher, T. N. & Thommen, D. S. Tertiary lymphoid structures in cancer. Science375, eabf9419 (2022). - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
