

A high-resolution meiotic crossover map from single-nucleus ATAC-seq reveals insights into the recombination landscape in mammals
- PMID: 40918063
- PMCID: PMC12408907
- DOI: 10.1093/nargab/lqaf122
A high-resolution meiotic crossover map from single-nucleus ATAC-seq reveals insights into the recombination landscape in mammals
Abstract
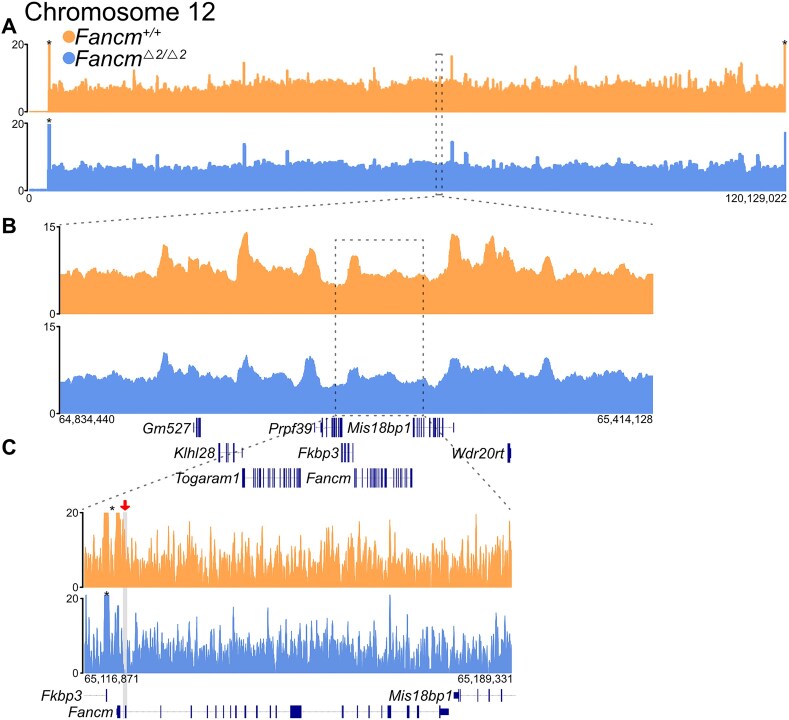
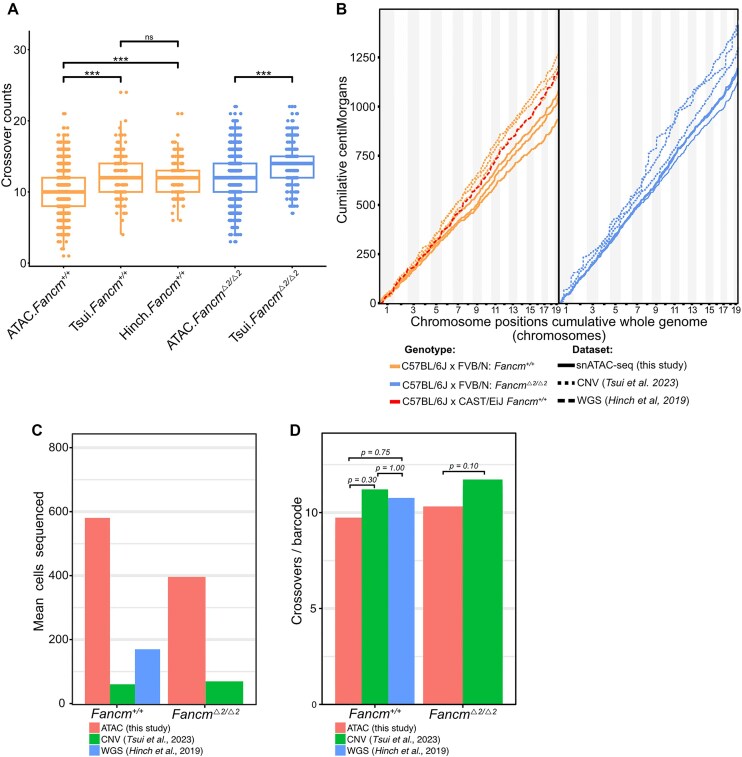
Meiotic crossovers promote correct chromosome segregation and the shuffling of genetic diversity. However, the measurement of crossovers remains challenging, impeding our ability to decipher the molecular mechanisms that are necessary for their formation and regulation. Here we demonstrate a novel repurposing of the single-nucleus Assay for Transposase Accessible Chromatin with sequencing (snATAC-seq) as a simple and high-throughput method to identify and characterize meiotic crossovers from haploid testis nuclei. We first validate the feasibility of obtaining genome-wide coverage from snATAC-seq by using ATAC-seq on bulk haploid mouse testis nuclei, ensuring adequate variant detection for haplotyping. Subsequently, we adapt droplet-based snATAC-seq for crossover detection, revealing >25 000 crossovers in F1 hybrid mice. Comparison between the wild type and a hyper-recombinogenic Fancm-deficient mutant mouse model confirmed an increase in crossover rates in this genotype, however with a distribution which was unchanged. We also find that regions with the highest rate of crossover formation are enriched for PRDM9. Our findings demonstrate the utility of snATAC-seq as a robust and scalable tool for high-throughput crossover detection, offering insights into meiotic crossover dynamics and elucidating the underlying molecular mechanisms. It is possible that the research presented here with snATAC-seq of haploid post-meiotic nuclei could be extended into fertility-related diagnostics.
© The Author(s) 2025. Published by Oxford University Press.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures






References
-
- Warren AC, Chakravarti A, Wong C et al. Evidence for reduced recombination on the nondisjoined chromosomes 21 in Down syndrome. Science. 1987; 237:652–4. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
