

Genome-Wide Insights Into the Genes and Pathways Shaping Human Foveal Development: Redefining the Genetic Landscape of Foveal Hypoplasia
- PMID: 40923693
- PMCID: PMC12425146
- DOI: 10.1167/iovs.66.12.22
Genome-Wide Insights Into the Genes and Pathways Shaping Human Foveal Development: Redefining the Genetic Landscape of Foveal Hypoplasia
Abstract
Purpose: To define the genetic architecture of foveal morphology and explore its relevance to foveal hypoplasia (FH), a hallmark of developmental macular disorders.
Methods: We applied deep-learning algorithms to quantify foveal pit depth from central optical coherence tomography (OCT) B-scans in 61,269 UK Biobank participants. A genome-wide association study (GWAS) was conducted using REGENIE, adjusting for age, sex, height, and ancestry. Rare coding variants (frequency <1%) were analyzed in an exome-wide rare-variant association study (RVAS). Candidate genes were prioritized using integrative mapping; pathway, cross-ancestry, and genetic-correlation analyses were exploratory.
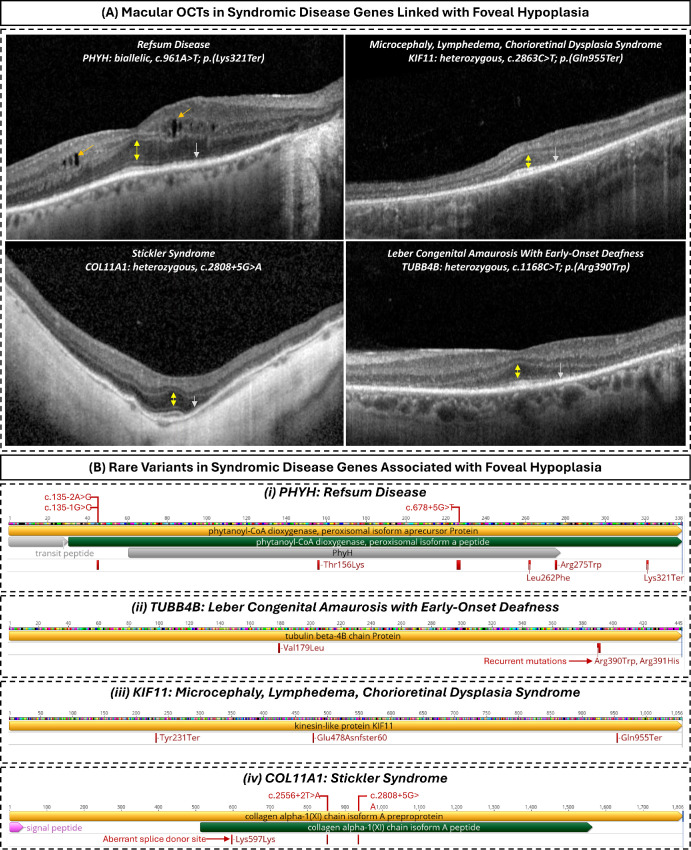
Results: GWAS identified 126 sentinel variants, including 47 novel associations. Integrative mapping prioritized 129 putative causal genes, with 64 not previously implicated in foveal biology. Enriched pathways included retinoic acid metabolism (e.g., CYP26A1), photoreceptor differentiation (e.g., VSX2), extracellular matrix organization, and pigmentation. RVAS identified missense variants in ACTN3 and ESYT3 (P < 5 × 10-⁹) associated with FH features. Polygenic scores were predictive across African and South Asian ancestries. Overlap was observed with monogenic FH genes (TYR, OCA2, PAX6, AHR) and with genes underlying systemic diseases (COL11A1, KIF11, TUBB4B, PHYH). Re-examination of OCTs in affected individuals confirmed FH in select cases, including those with recurrent TUBB4B p.(Arg390Trp) variants.
Conclusions: This is the first GWAS of human foveal morphology. Our findings redefine the genetic and biological framework underlying normal foveal development and foveal hypoplasia (FH). By linking common variation to rare monogenic disease, we establish a continuum model of FH with implications for future mechanistic and clinical investigation.
Conflict of interest statement
Disclosure:
Figures




Update of
-
Genome-Wide Insights into the Genes and Pathways Shaping Human Foveal Development.medRxiv [Preprint]. 2025 Jun 20:2025.06.18.25329836. doi: 10.1101/2025.06.18.25329836. medRxiv. 2025. Update in: Invest Ophthalmol Vis Sci. 2025 Sep 2;66(12):22. doi: 10.1167/iovs.66.12.22. PMID: 40585125 Free PMC article. Updated. Preprint.
References
MeSH terms
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
