

DNA methylation cooperates with genomic alterations during non-small cell lung cancer evolution
- PMID: 40931149
- PMCID: PMC12425823
- DOI: 10.1038/s41588-025-02307-x
DNA methylation cooperates with genomic alterations during non-small cell lung cancer evolution
Abstract
Aberrant DNA methylation has been described in nearly all human cancers, yet its interplay with genomic alterations during tumor evolution is poorly understood. To explore this, we performed reduced representation bisulfite sequencing on 217 tumor and matched normal regions from 59 patients with non-small cell lung cancer from the TRACERx study to deconvolve tumor methylation. We developed two metrics for integrative evolutionary analysis with DNA and RNA sequencing data. Intratumoral methylation distance quantifies intratumor DNA methylation heterogeneity. MR/MN classifies genes based on the rate of hypermethylation at regulatory (MR) versus nonregulatory (MN) CpGs to identify driver genes exhibiting recurrent functional hypermethylation. We identified DNA methylation-linked dosage compensation of essential genes co-amplified with neighboring oncogenes. We propose two complementary mechanisms that converge for copy number alteration-affected chromatin to undergo the epigenetic equivalent of an allosteric activity transition. Hypermethylated driver genes under positive selection may open avenues for therapeutic stratification of patients.
© 2025. The Author(s).
Conflict of interest statement
Competing interests: E.L.C. is currently employed by and holds shares in Achilles Therapeutics. N.K. acknowledges grant support from AstraZeneca. C.S. acknowledges grants from AstraZeneca, Boehringer Ingelheim, Bristol Myers Squibb, Pfizer, Roche-Ventana, Invitae (previously Archer Dx, a collaboration in minimal residual disease sequencing technologies), Ono Pharmaceutical and Personalis. He is Chief Investigator for the AZ MeRmaiD 1 and 2 clinical trials and is the Steering Committee Chair. He is also Co-Chief Investigator of the NHS Galleri trial funded by GRAIL and a paid member of GRAIL’s Scientific Advisory Board (SAB). He receives consultant fees from Achilles Therapeutics (he is also a SAB member), Bicycle Therapeutics (he is also a SAB member), Genentech, Medicxi, the China Innovation Centre of Roche, formerly the Roche Innovation Centre, Metabomed (until July 2022) and the Sarah Cannon Research Institute. C.S. has received honoraria from Amgen, AstraZeneca, Bristol Myers Squibb, GlaxoSmithKline, Illumina, MSD, Novartis, Pfizer and Roche-Ventana. C.S. has previously held stock options in Apogen Biotechnologies and GRAIL, and currently has stock options in Epic Bioscience and Bicycle Therapeutics, and has stock options and is co-founder of Achilles Therapeutics. C.S. declares a patent application (no. PCT/US2017/028013) for methods to lung cancer; for targeting neoantigens (no. PCT/EP2016/059401); for identifying patent response to immune checkpoint blockade (no. PCT/EP2016/071471); for determining HLA LOH (no. PCT/GB2018/052004); for predicting the survival rates of patients with cancer (no. PCT/GB2020/050221); for identifying patients who respond to cancer treatment (no. PCT/GB2018/051912); and for methods for lung cancer detection (no. US20190106751A1). C.S. is an inventor on a European patent application (no. PCT/GB2017/053289) relating to assay technology to detect tumor recurrence. This patent has been licensed to a commercial entity; under their terms of employment, C.S. is due a revenue share of any revenue generated from such license(s). J.D. has consulted for AvH. M.J.-H. has received funding from CRUK, the National Institutes of Health National Cancer Institute, the International Association for the Study of Lung Cancer, the Lung Cancer Research Foundation, the Rosetrees Trust, the UK and Ireland Neuroendocrine Tumour Society and the NIHR. M.J.-H. has consulted for, and is a member of, the Achilles Therapeutics SAB and Steering Committee, has received speaker honoraria from Pfizer, Astex Pharmaceuticals, the Oslo Cancer Cluster and Bristol Myers Squibb, and is listed as a co-inventor on a European patent application relating to methods to detect lung cancer (no. PCT/US2017/028013). This patent has been licensed to commercial entities and, under the terms of employment, M.J.-H. is due a share of any revenue generated from such license(s). A.M.F. is a co-inventor on a patent application to determine methods and systems for tumor monitoring (no. PCT/EP2022/077987). S.V. is a co-inventor on a patent of methods for detecting molecules in a sample (no. 10578620). The other authors declare no competing interests.
Figures
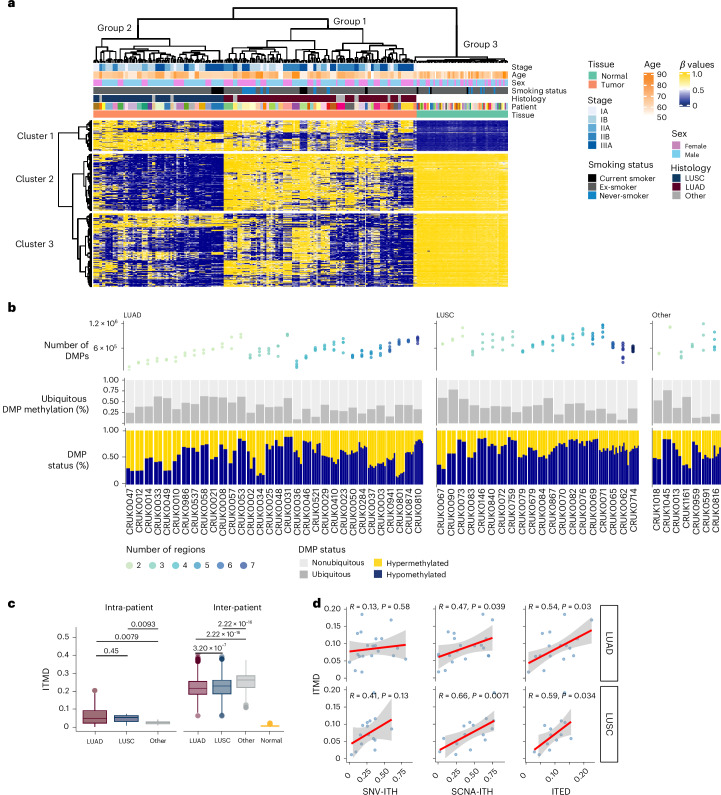
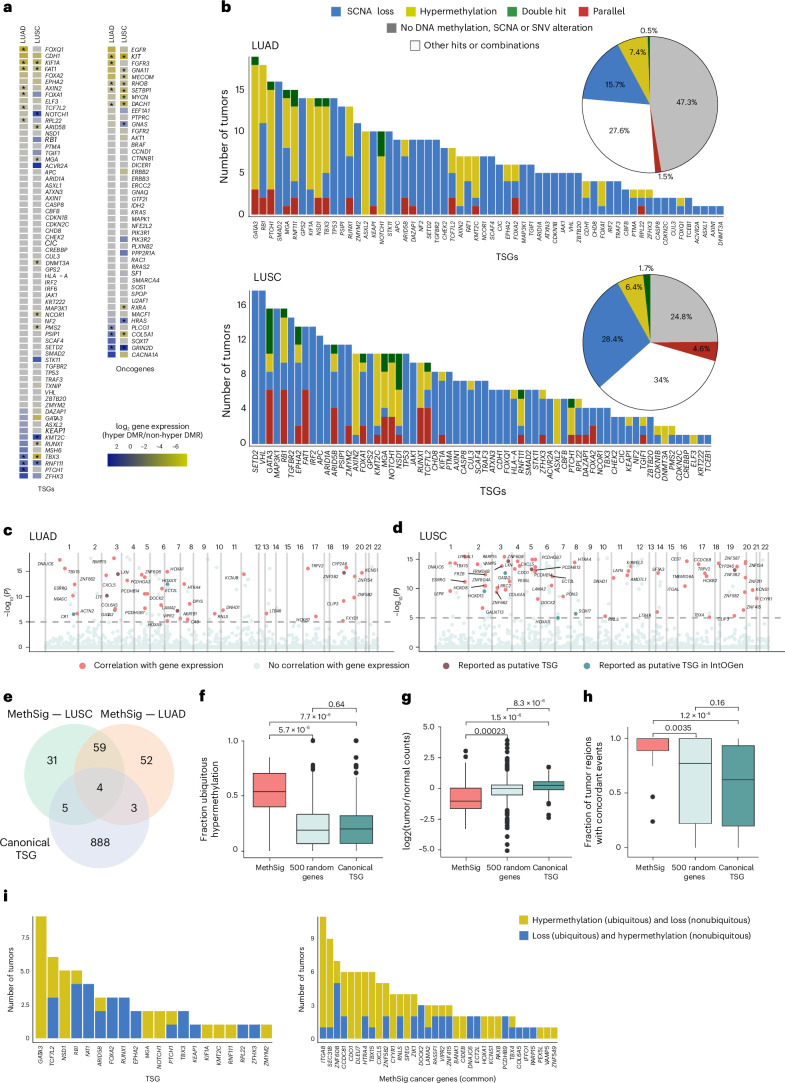

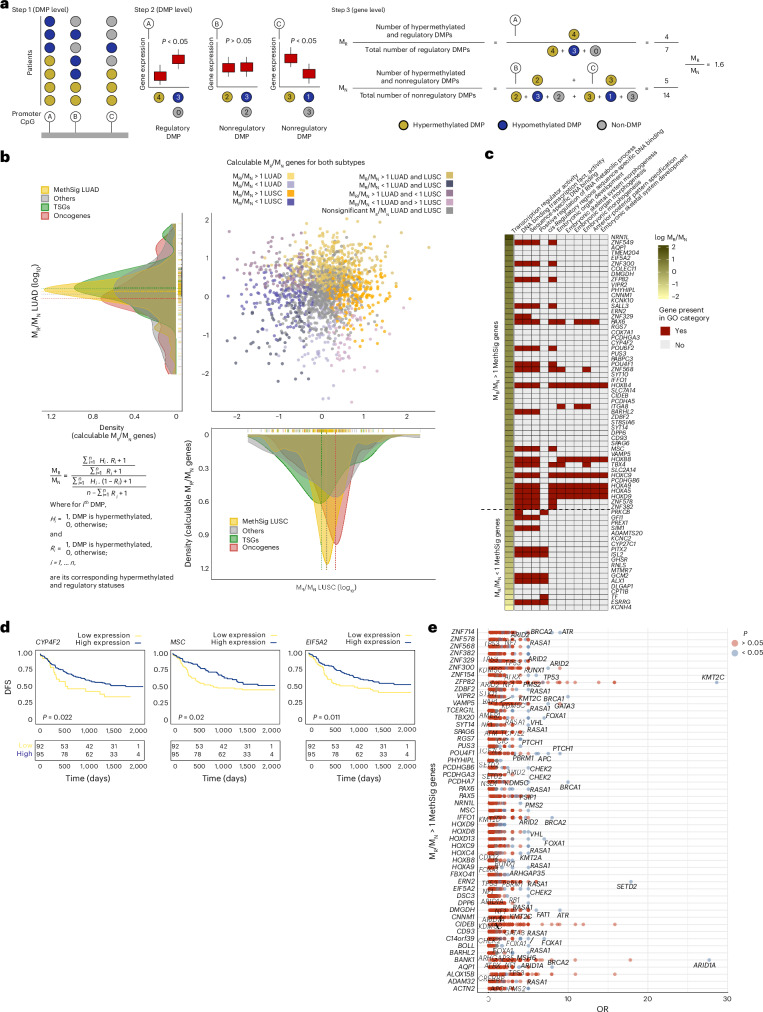








References
-
- Sung, H. et al. Global Cancer Statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin.71, 209–249 (2021). - PubMed
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
