

Categorizing prediction modes within low-pLDDT regions of AlphaFold2 structures: near-predictive, pseudostructure and barbed wire
- PMID: 40937679
- PMCID: PMC12485489
- DOI: 10.1107/S2059798325007843
Categorizing prediction modes within low-pLDDT regions of AlphaFold2 structures: near-predictive, pseudostructure and barbed wire
Abstract
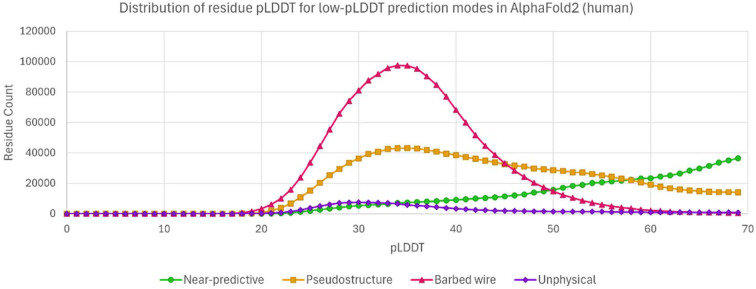
AlphaFold2 protein structure predictions are widely available for structural biology uses. These predictions, especially for eukaryotic proteins, frequently contain extensive regions predicted below the pLDDT = 70 level, the rule-of-thumb cutoff for high confidence. This work identifies major modes of behavior within low-pLDDT regions through a survey of human proteome predictions provided by the AlphaFold Protein Structure Database. The near-predictive mode resembles folded protein and can be a nearly accurate prediction. Barbed wire is extremely unprotein-like, being recognized by wide looping coils, an absence of packing contacts and numerous signature validation outliers, and it represents a region where the conformation has no predictive value. Pseudostructure presents an intermediate behavior with a misleading appearance of isolated and badly formed secondary-structure-like elements. These prediction modes are compared with annotations of disorder from MobiDB, showing general correlation between barbed wire/pseudostructure and many measures of disorder, an association between pseudostructure and signal peptides, and an association between near-predictive and regions of conditional folding. To enable users to identify these regions within a prediction, a new Phenix tool is developed encompassing the results of this work, including prediction annotation, visual markup and residue selection based on these prediction modes. This tool will help users develop expertise in interpreting difficult AlphaFold predictions and identify the near-predictive regions that can aid in molecular replacement when a prediction does not contain enough high-pLDDT regions.
Keywords: AlphaFold2; barbed wire; conditional folding; intrinsically disordered regions; low pLDDT; near-predictive; signal peptides; structure prediction; structure validation.
open access.
Conflict of interest statement
The authors declare no conflicts of interest.
Figures







Update of
-
Categorizing prediction modes within low-pLDDT regions of AlphaFold2 structures.bioRxiv [Preprint]. 2025 Jun 7:2025.06.06.658382. doi: 10.1101/2025.06.06.658382. bioRxiv. 2025. Update in: Acta Crystallogr D Struct Biol. 2025 Oct 1;81(Pt 10):558-572. doi: 10.1107/S2059798325007843. PMID: 40501571 Free PMC article. Updated. Preprint.
References
-
- Abramson, J., Adler, J., Dunger, J., Evans, R., Green, T., Pritzel, A., Ronneberger, O., Willmore, L., Ballard, A. J., Bambrick, J., Bodenstein, S. W., Evans, D. A., Hung, C. C., O’Neill, M., Reiman, D., Tunyasuvunakool, K., Wu, Z., Žemgulytė, A., Arvaniti, E., Beattie, C., Bertolli, O., Bridgland, A., Cherepanov, A., Congreve, M., Cowen-Rivers, A. I., Cowie, A., Figurnov, M., Fuchs, F. B., Gladman, H., Jain, R., Khan, Y. A., Low, C. M. R., Perlin, K., Potapenko, A., Savy, P., Singh, S., Stecula, A., Thillaisundaram, A., Tong, C., Yakneen, S., Zhong, E. D., Zielinski, M., Žídek, A., Bapst, V., Kohli, P., Jaderberg, M., Hassabis, D. & Jumper, J. M. (2024). Nature, 630, 493–500.
-
- Adzhubei, A. A., Sternberg, M. J. & Makarov, A. A. (2013). J. Mol. Biol.425, 2100–2132. - PubMed
-
- Baek, M., DiMaio, F., Anishchenko, I., Dauparas, J., Ovchinnikov, S., Lee, G. R., Wang, J., Cong, Q., Kinch, L. N., Schaeffer, R. D., Millán, C., Park, H., Adams, C., Glassman, C. R., DeGiovanni, A., Pereira, J. H., Rodrigues, A. V., van Dijk, A. A., Ebrecht, A. C., Opperman, D. J., Sagmeister, T., Buhlheller, C., Pavkov-Keller, T., Rathinaswamy, M. K., Dalwadi, U., Yip, C. K., Burke, J. E., Garcia, K. C., Grishin, N. V., Adams, P. D., Read, R. J. & Baker, D. (2021). Science, 373, 871–876.
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
