

This is a preprint.
Logan: Planetary-Scale Genome Assembly Surveys Life's Diversity
- PMID: 40950083
- PMCID: PMC12424806
- DOI: 10.1101/2024.07.30.605881
Logan: Planetary-Scale Genome Assembly Surveys Life's Diversity
Abstract
The breadth of life's diversity is unfathomable, but public nucleic acid sequencing data offers a window into the dispersion and evolution of genetic diversity across Earth. However the rapid growth and accumulation of sequence data have outpaced efficient analysis capabilities. The largest collection of freely available sequencing data is the Sequence Read Archive (SRA), comprising 27.3 million datasets or 5 × 1016 basepairs. To realize the potential of the SRA, we constructed Logan, a massive sequence assembly transforming short reads into long contigs and compressing the data over 100-fold, enabling highly efficient petabase-scale analysis. We created Logan-Search, a k-mer index of Logan for free planetary-scale sequence search, returning matches in minutes. We used Logan contigs to identify >200 million plastic-degrading enzyme homologs, and validate novel enzymes with catalytic activities exceeding current reference standards. Further, we vastly expand the known diversity of proteins (30-fold over UniRef50), plasmids (22-fold over PLSDB), P4 satellites (4.5-fold), and the recently described Obelisk RNA elements (3.7-fold). Logan also enables ecological and biomedical data mining, such as global tracking of antimicrobial resistance genes and the characterization of viral reactivation across millions of human BioSamples. By transforming the SRA, Logan democratizes access to the world's public genetic data and opens frontiers in biotechnology, molecular ecology, and global health.
Conflict of interest statement
8Competing interests The authors declare no competing interests.
Figures


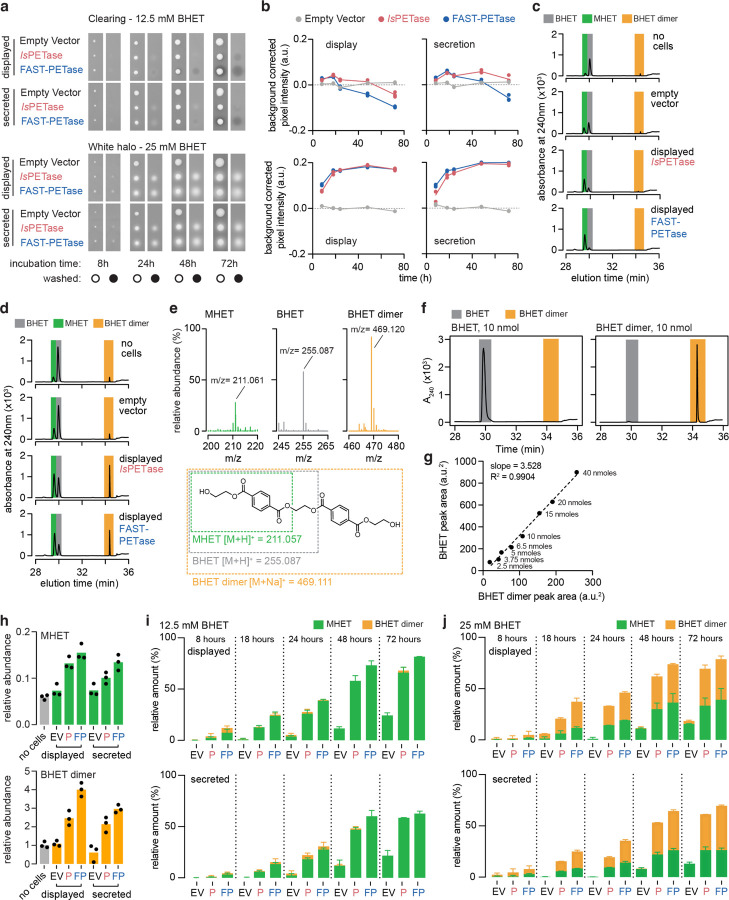






References
-
- Edgar Robert C, Taylor Brie, Lin Victor, Altman Tomer, Barbera Pierre, Meleshko Dmitry, Lohr Dan, Novakovsky Gherman, Buchfink Benjamin, Al-Shayeb Basem, et al. Petabase-scale sequence alignment catalyses viral discovery. Nature, 602(7895):142–147, 2022. - PubMed
-
- Bradley Phelim, Den Bakker Henk C, Rocha Eduardo PC, McVean Gil, and Iqbal Zamin. Ultrafast search of all deposited bacterial and viral genomic data. Nature Biotechnology, 37(2):152–159, 2019.
-
- Karasikov Mikhail, Mustafa Harun, Danciu Daniel, Barber Christopher, Zimmermann Marc, Rätsch Gunnar, and Kahles André. Metagraph: Indexing and analysing nucleotide archives at petabase-scale. BioRxiv, pages 2020–10, 2020.
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous