

Population-genomics reveals a dual ancestry of grizzly bears
- PMID: 40950719
- PMCID: PMC12432854
- DOI: 10.1016/j.isci.2025.112870
Population-genomics reveals a dual ancestry of grizzly bears
Abstract
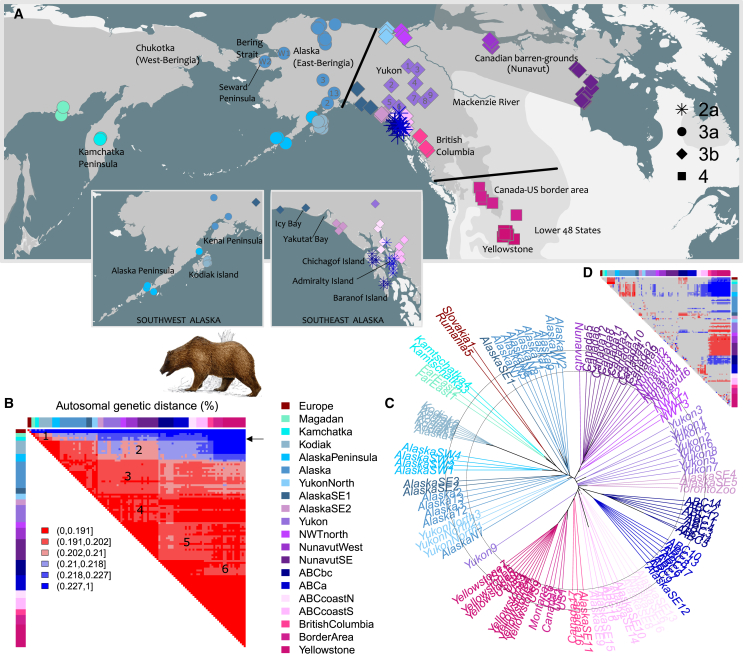
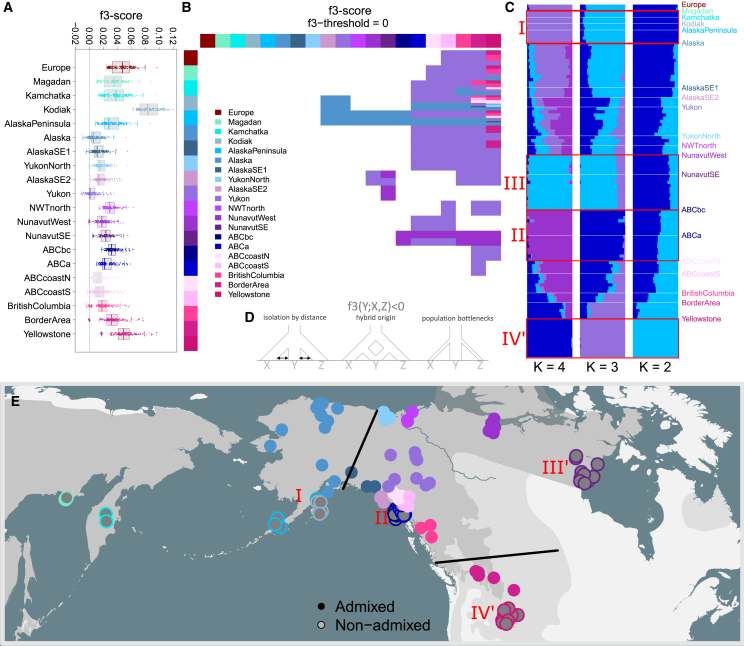
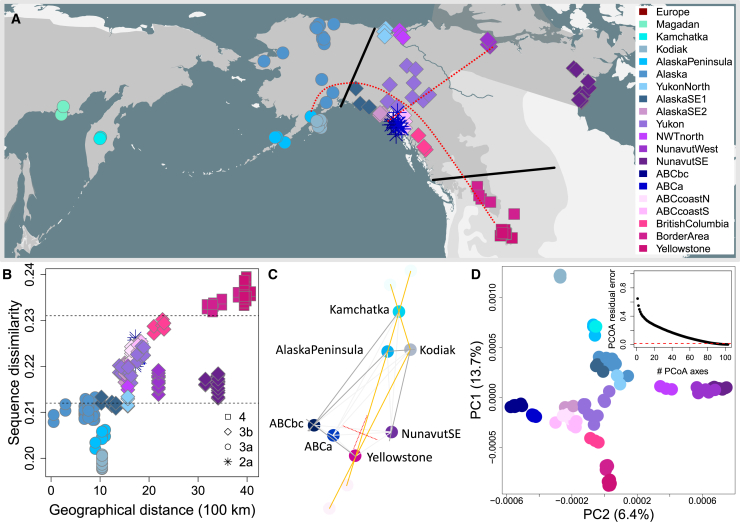
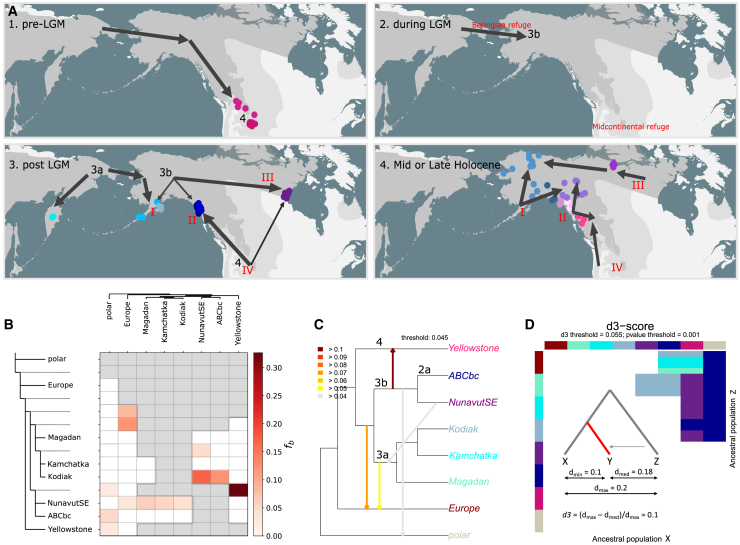
Genetic variation among populations reflects both past demographic events and current population connectivity. We investigate the primary drivers of genetic differentiation in American brown bears (Ursus arctos) using 108 nuclear genomes. Our analyses reveal that genome-wide distances conform to neither an isolation-by-distance model nor a bifurcating tree structure. Building on previous ancient-DNA and fossil studies, we propose a demographic scenario in which continent-wide admixture during the Late Holocene has obscured, but did not erase, the genetic legacy of earlier colonization waves and subsequent gene flow events. The most persistent signals of these past events are striking genetic similarities between populations now separated by water barriers, including Kamchatka and Southwest Alaska bears. Our findings underscore that convergence to migration-drift equilibrium takes time, making genetic distance an imperfect proxy for present-day population connectivity.
Keywords: Animals; Biological classification; Evolutionary history; Phylogenetics.
© 2025 The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures



References
LinkOut - more resources
Full Text Sources
Miscellaneous
