

Smooth muscle expression of RNA editing enzyme ADAR1 controls activation of the RNA sensor MDA5 in atherosclerosis
- PMID: 40958051
- PMCID: PMC12520979
- DOI: 10.1038/s44161-025-00710-5
Smooth muscle expression of RNA editing enzyme ADAR1 controls activation of the RNA sensor MDA5 in atherosclerosis
Abstract
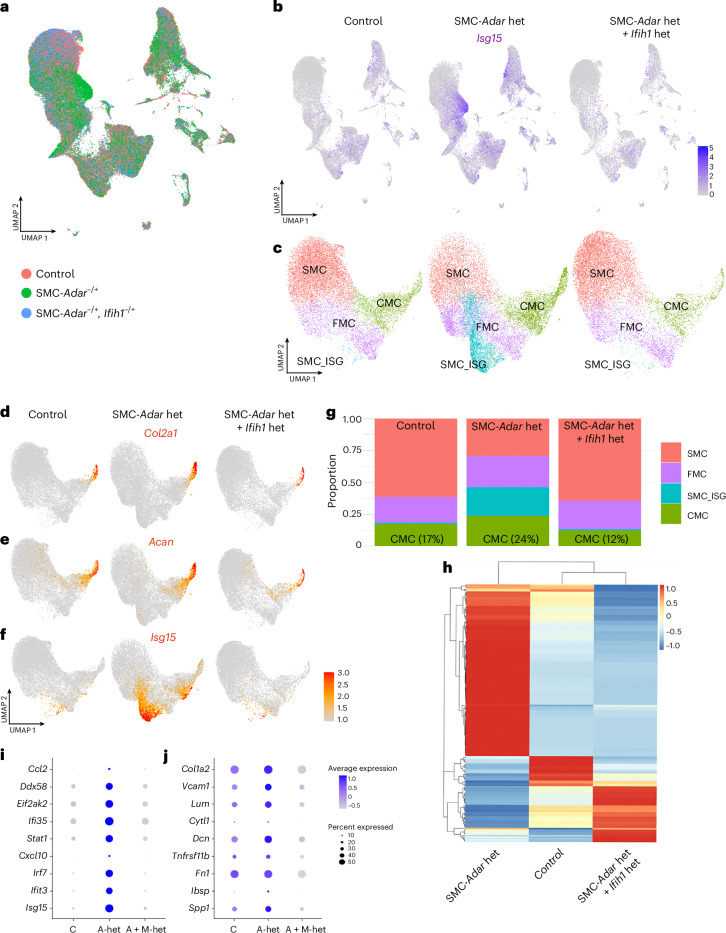
Although genetic risk in coronary artery disease (CAD) is linked to changes in gene expression, recent discoveries have revealed a major role for A-to-I RNA editing in CAD. ADAR1 edits immunogenic double-stranded RNA (dsRNA), preventing activation of the dsRNA sensor MDA5 (IFIH1) and downstream interferon-stimulated gene signaling. Using human plaque analysis and human coronary artery smooth muscle cells (SMCs), here, we show that SMCs uniquely require RNA editing and that MDA5 activation regulates SMC phenotype. In a conditional SMC-specific Adar deletion mouse model on an atherosclerosis-prone background, combined with Ifih1 deletion and single-cell RNA sequencing, we demonstrate that ADAR1 preserves vascular integrity and limits atherosclerosis and calcification by suppressing MDA5 activation. Analysis of the Athero-Express carotid endarterectomy cohort further shows that interferon-stimulated gene expression correlates with SMC modulation, plaque instability and calcification. These findings reveal a fundamental mechanism of CAD, where cell type and context-specific RNA editing modulates genetic risk and vascular disease progression.
© 2025. The Author(s).
Conflict of interest statement
Competing interests: C.S.W. is a consultant for AiRNA Bio and Avidity Biosciences. T.Q. serves on the scientific advisory board for Amgen. J.B.L. is a cofounder of AIRNA Bio and a consultant for Risen Pharma. J.B.L. and Q.L. are named inventors of a patent filed by Stanford University (WO/2023/239781), describing a method related to genetic prediction of RNA editing that relates to human prediction of dsRNA burden. S.W.v.d.L. has received Roche funding for unrelated work. The other authors declare no competing interests.
Figures

















Update of
-
Smooth muscle expression of RNA editing enzyme ADAR1 controls activation of RNA sensor MDA5 in atherosclerosis.bioRxiv [Preprint]. 2025 Jun 25:2024.07.08.602569. doi: 10.1101/2024.07.08.602569. bioRxiv. 2025. Update in: Nat Cardiovasc Res. 2025 Oct;4(10):1241-1257. doi: 10.1038/s44161-025-00710-5. PMID: 39026721 Free PMC article. Updated. Preprint.
References
MeSH terms
Substances
Grants and funding
- R01MH115080/U.S. Department of Health & Human Services | NIH | National Institute of Mental Health (NIMH)
- R35GM144100/U.S. Department of Health & Human Services | NIH | National Institute of General Medical Sciences (NIGMS)
- K08 HL167699/HL/NHLBI NIH HHS/United States
- L30 HL159413/HL/NHLBI NIH HHS/United States
- R01HL139478/U.S. Department of Health & Human Services | NIH | National Heart, Lung, and Blood Institute (NHLBI)
- R01 HL151535/HL/NHLBI NIH HHS/United States
- R01GM102484/U.S. Department of Health & Human Services | NIH | National Institute of General Medical Sciences (NIGMS)
- UM1 HG011972/HG/NHGRI NIH HHS/United States
- K08HL167699/U.S. Department of Health & Human Services | NIH | National Heart, Lung, and Blood Institute (NHLBI)
- F32 HL165854/HL/NHLBI NIH HHS/United States
- R01 HL145708/HL/NHLBI NIH HHS/United States
- U01DK142283-01/U.S. Department of Health & Human Services | NIH | National Institute of Diabetes and Digestive and Kidney Diseases (National Institute of Diabetes & Digestive & Kidney Diseases)
- 20CDA35310303/American Heart Association (American Heart Association, Inc.)
- R00HL150319/U.S. Department of Health & Human Services | NIH | National Heart, Lung, and Blood Institute (NHLBI)
- 24POST1187860/American Heart Association (American Heart Association, Inc.)
- R00 HL150319/HL/NHLBI NIH HHS/United States
- 23POST1018991/American Heart Association (American Heart Association, Inc.)
- 23CDA1042900/American Heart Association (American Heart Association, Inc.)
- K08HL153798/U.S. Department of Health & Human Services | NIH | National Heart, Lung, and Blood Institute (NHLBI)
- R01HL134817/U.S. Department of Health & Human Services | NIH | National Heart, Lung, and Blood Institute (NHLBI)
- U01 DK142283/DK/NIDDK NIH HHS/United States
- R35 GM144100/GM/NIGMS NIH HHS/United States
- R01 MH115080/MH/NIMH NIH HHS/United States
- K08HL177251/U.S. Department of Health & Human Services | NIH | National Heart, Lung, and Blood Institute (NHLBI)
- L30HL159413/U.S. Department of Health & Human Services | NIH | National Heart, Lung, and Blood Institute (NHLBI)
- 24CDA1272805/American Heart Association (American Heart Association, Inc.)
- F32HL160067/U.S. Department of Health & Human Services | NIH | National Heart, Lung, and Blood Institute (NHLBI)
- 24SCEFIA1248386/American Heart Association (American Heart Association, Inc.)
- R01HL179083/U.S. Department of Health & Human Services | NIH | National Heart, Lung, and Blood Institute (NHLBI)
- R01 HL139478/HL/NHLBI NIH HHS/United States
- R01 HL179083/HL/NHLBI NIH HHS/United States
- R01HL151535/U.S. Department of Health & Human Services | NIH | National Heart, Lung, and Blood Institute (NHLBI)
- R01HL156846/U.S. Department of Health & Human Services | NIH | National Heart, Lung, and Blood Institute (NHLBI)
- R01 HL134817/HL/NHLBI NIH HHS/United States
- K08 HL177251/HL/NHLBI NIH HHS/United States
- R01 GM102484/GM/NIGMS NIH HHS/United States
- R01HL145708/U.S. Department of Health & Human Services | NIH | National Heart, Lung, and Blood Institute (NHLBI)
- F32 HL160067/HL/NHLBI NIH HHS/United States
- R01 HL156846/HL/NHLBI NIH HHS/United States
- K08 HL153798/HL/NHLBI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Miscellaneous