

Focal Adhesion Kinase Intersects With the BRD4-MYC Axis and YAP1 to Drive Tumor Cell Growth, Phenotypic Plasticity, Stemness, and Metastatic Potential in Colorectal Cancer
- PMID: 40959971
- PMCID: PMC12441738
- DOI: 10.1002/cam4.71227
Focal Adhesion Kinase Intersects With the BRD4-MYC Axis and YAP1 to Drive Tumor Cell Growth, Phenotypic Plasticity, Stemness, and Metastatic Potential in Colorectal Cancer
Abstract
Objective: Colorectal cancer (CRC) remains one of the leading causes of cancer-related death worldwide due to the lack of effective therapies. Here we explored the clinical basis and therapeutic promise of the integrin-focal adhesion kinase (FAK)-dependent pathway for CRC.
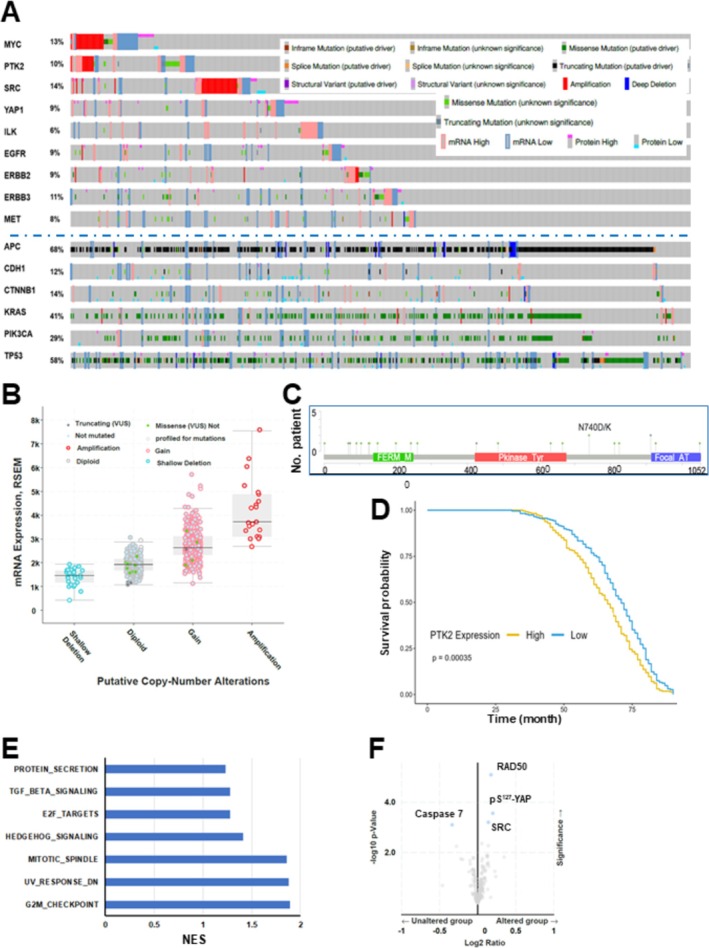
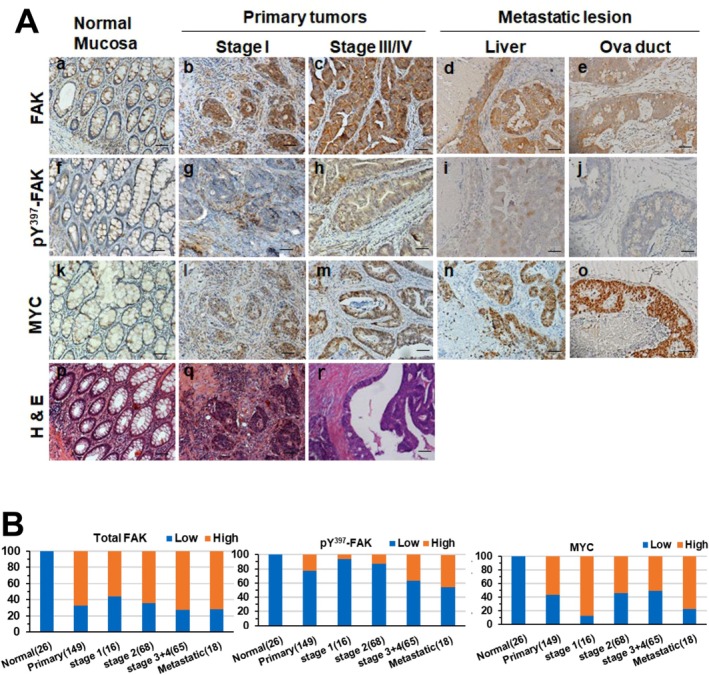
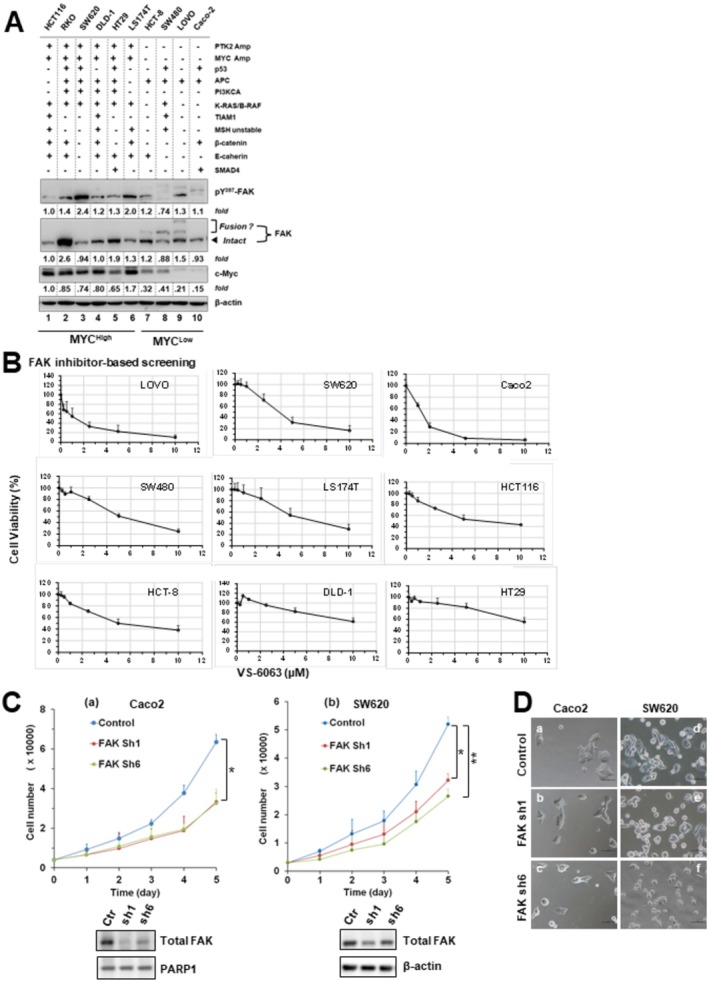
Methods and results: Our bioinformatic and histological analyses showed that FAK was markedly upregulated at both mRNA and protein and signaling levels in the two CRC patient cohorts. Particularly, the portion of carcinomas carrying active FAK (Y397phosphorylation) increased by threefold from stage I to III/IV tumors or metastatic lesions. Consistent with this clinic landscape, FAK inhibition via knockdown or chemical inhibitors suppressed tumor cell growth largely in the subset of CRC cell lines with low MYC expression. In contrast, the FAK inhibition was less effective in the cell line pool with high MYC expression. The resistance to FAK targeting diminished upon a co-inhibition of BRD4 via BET inhibitors. It coincided with an induction of cell cycle arrest at G1-S and G2-M phases, elevated apoptosis and chemosensitivity (paclitaxel and oxaliplatin), and impaired stemness. Mechanistically, the BET inhibitor induced an EMT-like phenotype, tilting tumor cell dependence toward the integrin-FAK axis. Moreover, inhibiting FAK alone or in combination with SRC or BRD4 markedly suppressed cell motility and the YAP or MYC activation, and restored the expression of the long isoform BRD4. Also, co-genomic/genetic dysregulations of FAK and YAP1 or SRC strongly correlated with poor disease-free patient survival.
Conclusion: Overall, our study highlights the potent pro-malignant role of the integrin-FAK axis in CRC, fueling its targeting as a single agent or synthetic lethal-based therapy.
Keywords: BRD4; FAK; MYC; colorectal cancer; metastasis; tumor growth.
© 2025 The Author(s). Cancer Medicine published by John Wiley & Sons Ltd.
Conflict of interest statement
The authors declare no conflicts of interest.
Figures




References
-
- Bray F., Laversanne M., Sung H., et al., “Global Cancer Statistics 2022: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries,” CA: A Cancer Journal for Clinicians 74, no. 3 (2024): 229–263. - PubMed
-
- Dienstmann R., Vermeulen L., Guinney J., Kopetz S., Tejpar S., and Tabernero J., “Consensus Molecular Subtypes and the Evolution of Precision Medicine in Colorectal Cancer,” Nature Reviews. Cancer 17, no. 4 (2017): 268. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
