

Integrative Genotyping and Analysis of Canine Structural Variation Using Long-read and Short-read Data
- PMID: 40971676
- PMCID: PMC12481690
- DOI: 10.1093/gbe/evaf173
Integrative Genotyping and Analysis of Canine Structural Variation Using Long-read and Short-read Data
Abstract
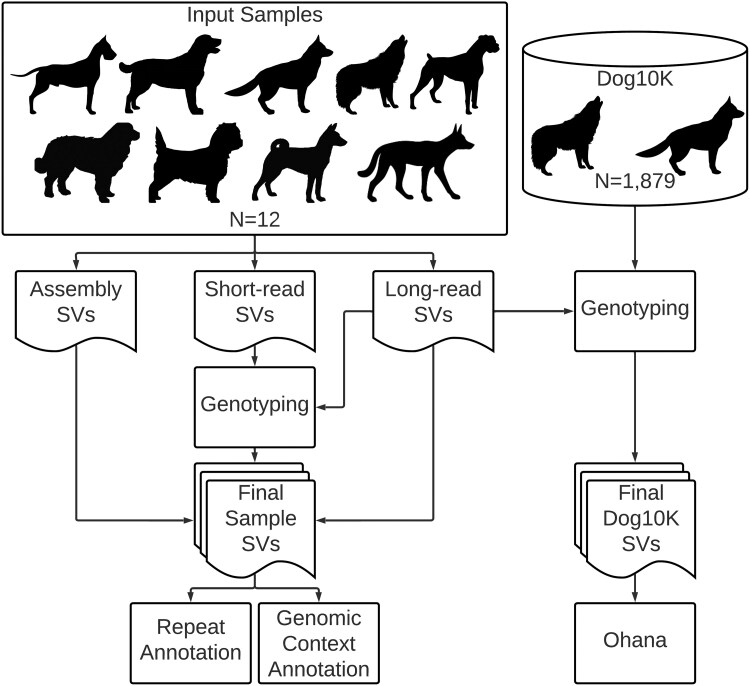
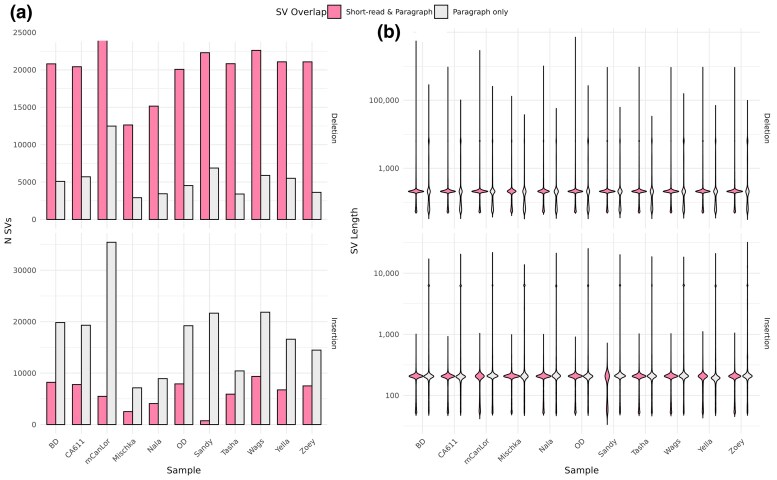
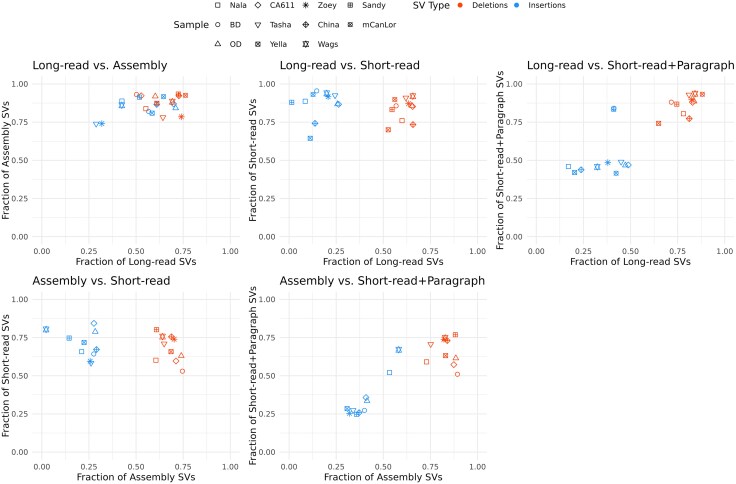
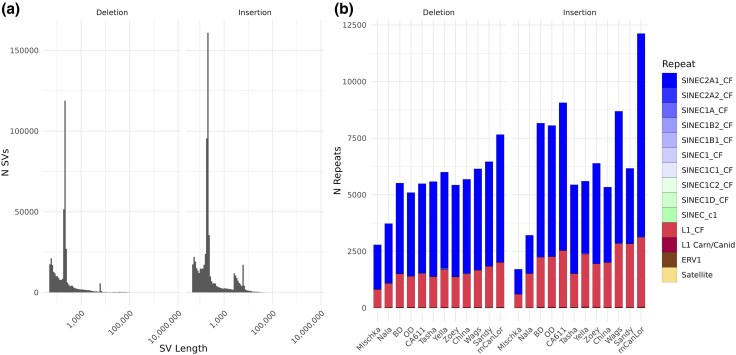
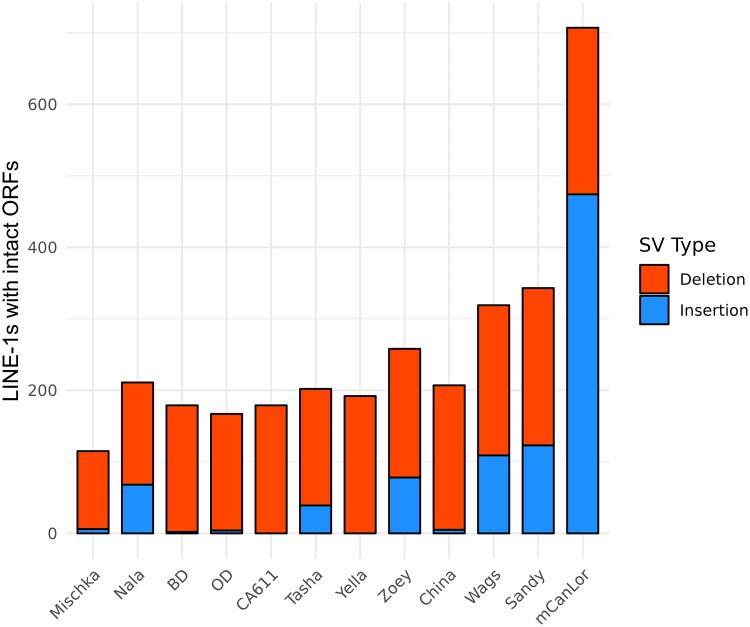
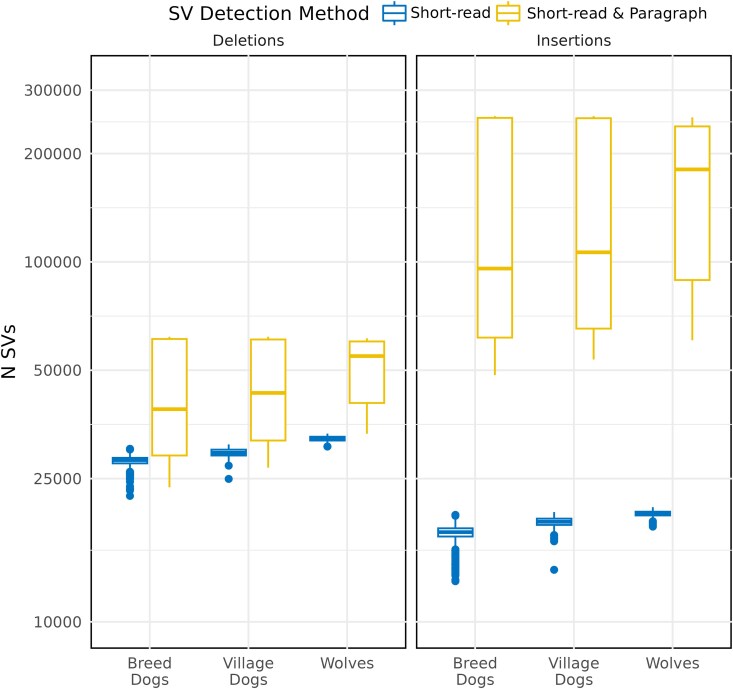
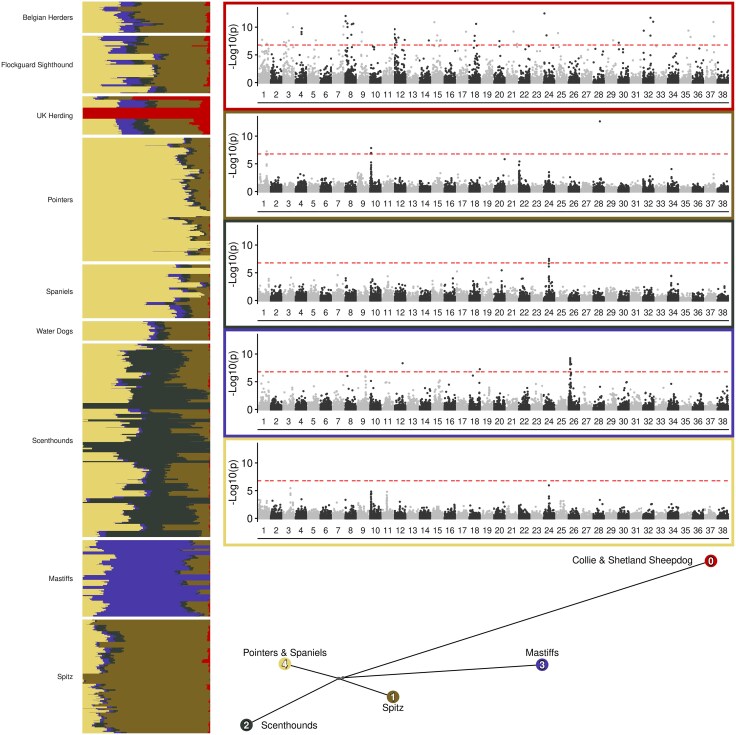
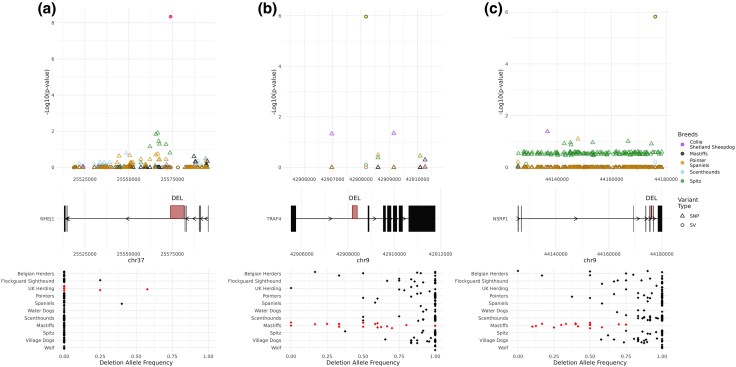
Structural variation makes an important contribution to canine evolution and phenotypic differences. Although recent advances in long-read sequencing have enabled the generation of multiple canine genome assemblies, most prior analyses of structural variation have relied on short-read sequencing. To offer a more complete assessment of structural variation in canines, we performed an integrative analysis of structural variants present in 12 canine samples with available long-read and short-read sequencing data along with genome assemblies. Use of long-reads permits the discovery of heterozygous variation that is absent in existing haploid assembly representations while offering a marked increase in the ability to identify insertion variants relative to short-read approaches. Examination of the size spectrum of structural variants shows that dimorphic LINE-1 and SINE variants account for over 45% of all deletions and identified 1,410 LINE-1s with intact open reading frames that show presence-absence dimorphism. Using a graph-based approach, we genotype newly discovered structural variants in an existing collection of 1,879 resequenced dogs and wolves, generating a variant catalog containing a 56.5% increase in the number of deletions and 705% increase in the number of insertions previously found in the analyzed samples. Examination of allele frequencies across admixture components present across breed clades identified 283 structural variants evolving with a signature of selection.
Keywords: long-read sequencing; mobile elements; structural variants.
© The Author(s) 2025. Published by Oxford University Press on behalf of Society for Molecular Biology and Evolution.
Figures

References
MeSH terms
LinkOut - more resources
Full Text Sources
