

Delineating the pathogenic threshold and phenotypic spectrum of SCA27B: findings from a large French-Canadian cohort
- PMID: 40974444
- PMCID: PMC12450228
- DOI: 10.1007/s00415-025-13387-4
Delineating the pathogenic threshold and phenotypic spectrum of SCA27B: findings from a large French-Canadian cohort
Abstract
Background: Autosomal dominant spinocerebellar ataxia 27B (SCA27B), caused by an intronic (GAA•TTC) repeat expansion in FGF14, is a common cause of late-onset cerebellar ataxia, but its genotypic and phenotypic spectrum remains to be fully established.
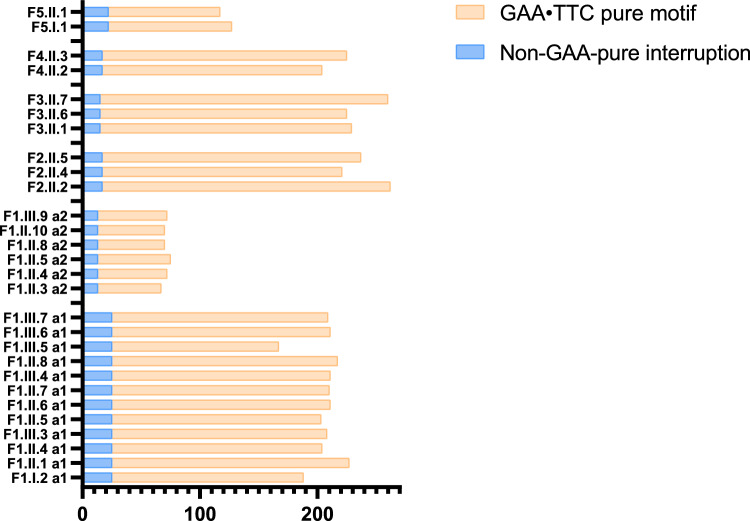
Methods: We analysed the FGF14 (GAA•TTC) repeat expansion in a cohort of 134 patients with ataxia and 822 controls from Quebec. We conducted segregation study in large families to further characterize intergenerational repeat instability.
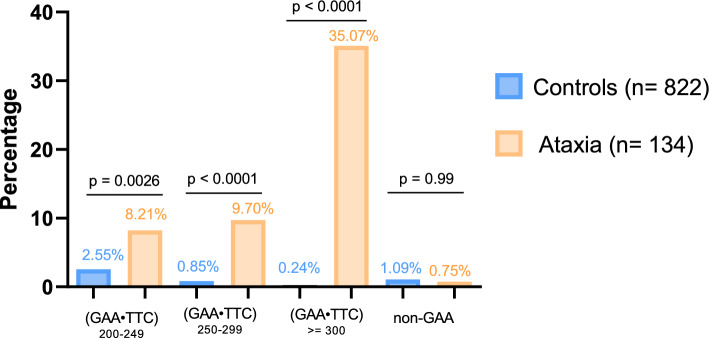
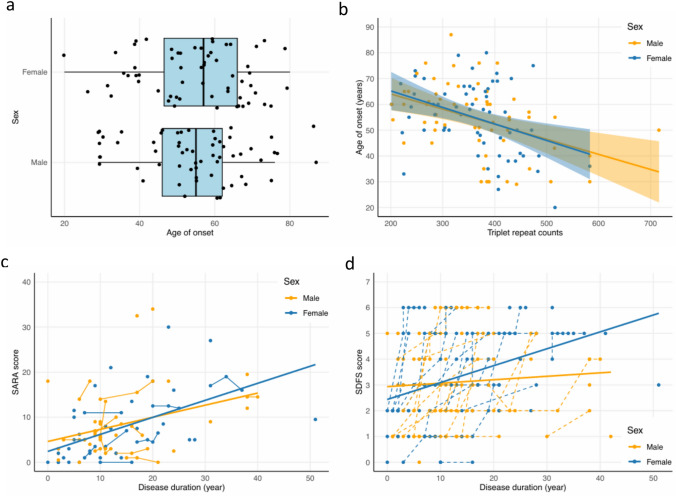
Results: We found a significant enrichment of (GAA•TTC)≥200 alleles in the ataxia cohort compared to controls (53.0%, 71/134, vs 3.6%, 30/822, p < 0.0001), including for (GAA•TTC)200-249 alleles (8.2% vs 2.6%, p = 0.0026). We identified 12 ataxic patients with a phenotype compatible with SCA27B carrying a (GAA•TTC)200-249 expansion supporting the pathogenicity of these alleles in some patients. We further delineated the phenotype of 125 symptomatic individuals from 69 families who carried an FGF14 (GAA•TTC)≥200 repeat expansion. Patients with (GAA•TTC)200-249, (GAA•TTC)250-299, and (GAA•TTC)≥300 had a similar phenotype. We observed that 14% of patients with episodic symptoms (13/92) had severe episodes that were initially misdiagnosed as stroke, vestibular neuritis, Wernicke's encephalopathy, or seizures.
Discussion and conclusion: This large cohort demonstrates that (GAA•TTC)200-249 alleles are enriched in patients with ataxia compared to controls and can be pathogenic for SCA27B, supporting the need to define a lower pathogenic threshold in the presence of specific clinical criteria.
Keywords: Ataxia; FGF14; French–Canadian; SCA27B.
© 2025. The Author(s).
Conflict of interest statement
Declarations. Conflicts of interest: Matthis Synofzik has received consultancy honoraria from Ionis, UCB, Prevail, Orphazyme, Biogen, Servier, Reata, GenOrph, AviadoBio, Biohaven, Zevra, Lilly, Quince, Neurocrine, and Solaxa, all unrelated to the present manuscript. Ethical approval: This study was performed in accordance with the ethical standards as laid down in the 1964 Declaration of Helsinki. The institutional review board of the Montreal Neurological Hospital (MPE-CUSM-15–915 and CHUM-ND02.045) approved this study and all participants provided written informed consent.
Figures




References
-
- Ashton C, Indelicato E, Pellerin D et al (2023) Spinocerebellar ataxia 27B: episodic symptoms and acetazolamide response in 34 patients. Brain Commun 5:1–5. 10.1093/braincomms/fcad239 - DOI
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
