

Functional mapping of epigenomic regulators uncovers coordinated tumor suppression by the HBO1 and MLL1 complexes
- PMID: 40997327
- PMCID: PMC12469823
- DOI: 10.1158/2159-8290.CD-24-1565
Functional mapping of epigenomic regulators uncovers coordinated tumor suppression by the HBO1 and MLL1 complexes
Abstract
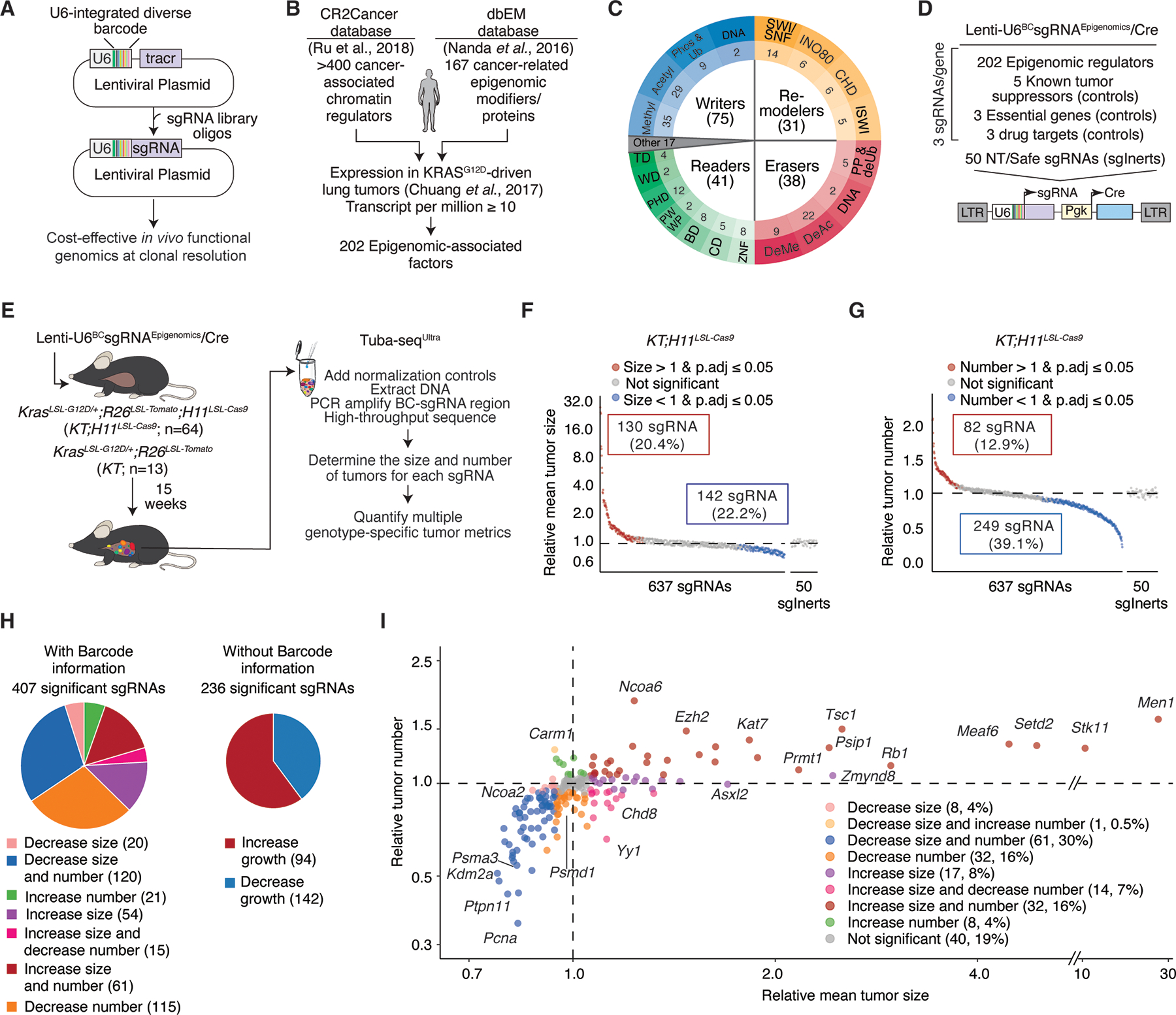
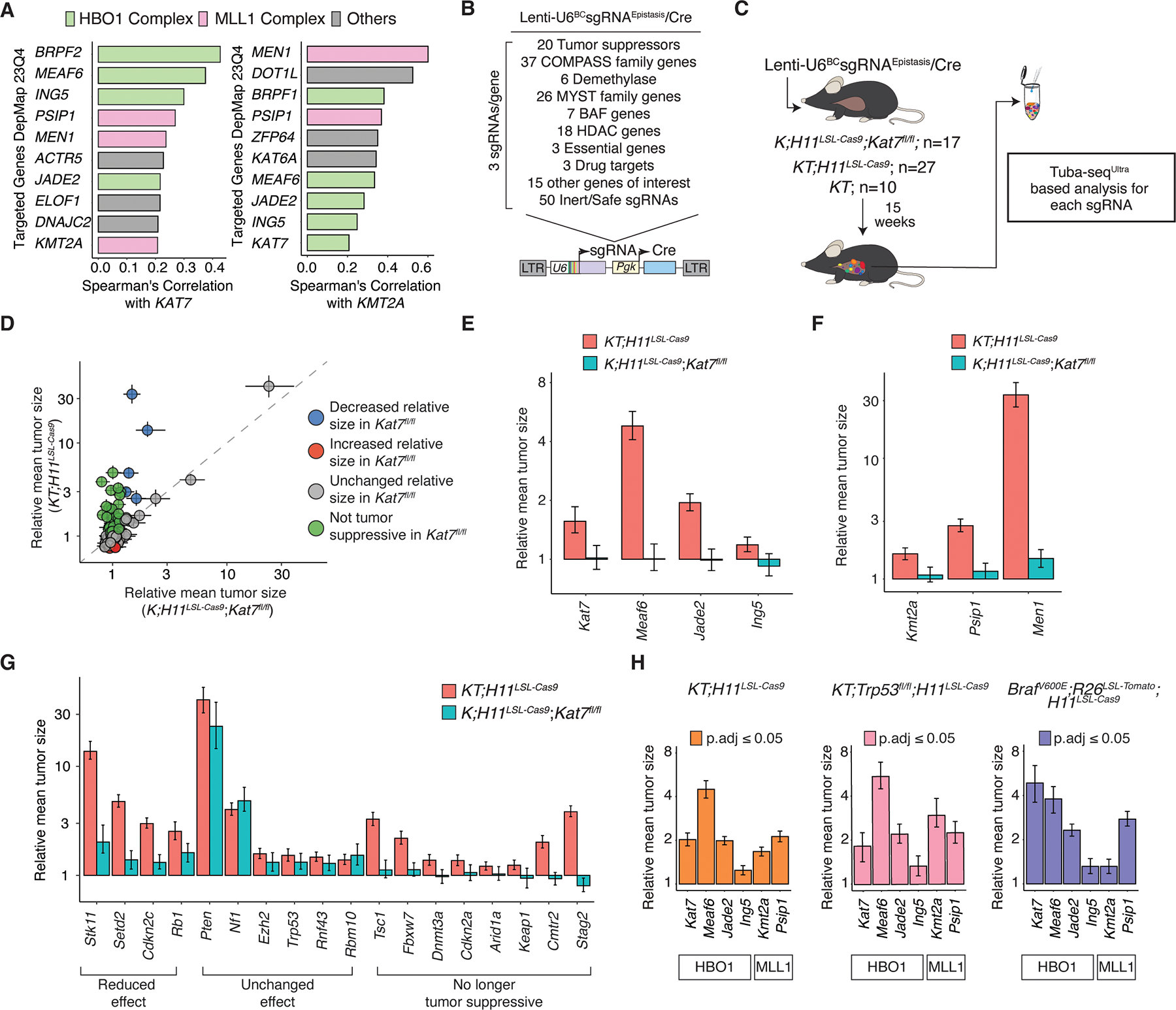
Epigenomic dysregulation is widespread in cancer. However, the specific epigenomic regulators and the processes they control to drive cancer phenotypes are poorly understood. We employed a novel high-throughput in vivo method to perform iterative functional screens of >250 epigenomic regulators within autochthonous oncogenic KRAS-driven lung tumors. We identified many previously unappreciated epigenomic tumor-suppressor and tumor-dependency genes. We show that a specific HBO1 complex and MLL1 complex are robust tumor suppressors in lung adenocarcinoma. Histone modifications generated by HBO1 complex are frequently reduced in human lung adenocarcinomas and are associated with worse clinical features. HBO1 and MLL1 complexes co-occupy shared genomic regions, impact chromatin accessibility, and control the expression of canonical tumor suppressor genes and lineage fidelity. The HBO1 complex is epistatic with the MLL1 complex and other tumor suppressor genes in lung adenocarcinoma development. Collectively, these results provide a phenotypic roadmap of epigenomic regulators in lung tumorigenesis in vivo.
Conflict of interest statement
Conflict of Interest Statement
M.M.W and D.A.P are founders and hold equity in Guide Oncology. The other authors declare no competing interest.
Figures





Update of
-
Functional mapping of epigenetic regulators uncovers coordinated tumor suppression by the HBO1 and MLL1 complexes.bioRxiv [Preprint]. 2024 Aug 20:2024.08.19.607671. doi: 10.1101/2024.08.19.607671. bioRxiv. 2024. Update in: Cancer Discov. 2025 Sep 26. doi: 10.1158/2159-8290.CD-24-1565. PMID: 39229041 Free PMC article. Updated. Preprint.
References
-
- Hanahan D Hallmarks of Cancer: New Dimensions. Cancer Discov 2022;12(1):31–46 doi 10.1158/2159-8290.CD-21-1059. - DOI - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
