

This is a preprint.
Comprehensive benchmarking of somatic structural variant detection at ultra-low allele fractions
- PMID: 41000761
- PMCID: PMC12458932
- DOI: 10.1101/2025.09.18.677206
Comprehensive benchmarking of somatic structural variant detection at ultra-low allele fractions
Abstract
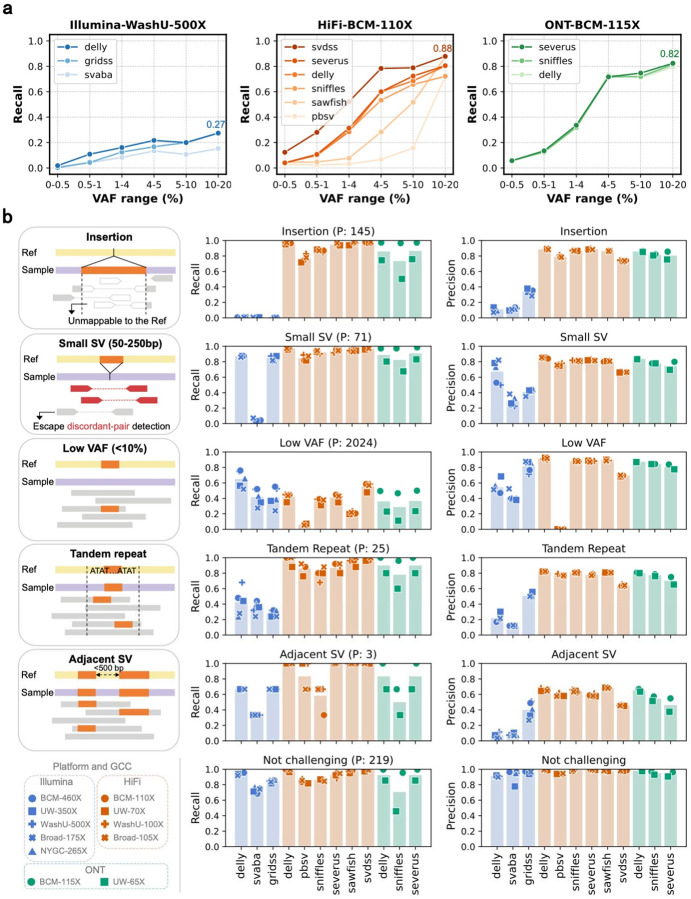
Postzygotic mosaicism gives rise to somatic structural variants (SVs) at ultra-low variant allele fractions (VAFs), which pose challenges for detection due to the high-coverage sequencing required and noise introduced by sequencing artifacts. Although somatic SV detection has been extensively studied in cancer, these studies are not directly applicable to the study of tissue mosaicism, as they rely on matched normals, target higher VAF ranges, and are enriched for different types of SVs. We present comprehensive benchmark data and best practices for non-cancer somatic SV detection. We created a synthetic mosaic sample by combining six HapMap individuals at varying proportions, generating allele fractions as low as 0.25%. This sample was sequenced to ~2,300x total coverage using Illumina, PacBio, and Nanopore technologies across multiple sequencing centers. A high-confidence benchmark SV set containing over 21,000 pseudo-somatic insertions and deletions ≥50bp was derived from haplotype-resolved assemblies. We evaluated 12 SV discovery pipelines and identified caller-specific strengths and sequencing platform-specific shortcomings. We find that short read-based approaches show reduced recall for insertions and repeat-associated SVs, whereas long-read sequencing achieves high accuracy throughout the genome, increasing linearly with coverage. The best algorithm's sensitivity exceeded 80% for VAFs ≥4% and 15% for VAFs of 0.5-1% with 60x coverage. The publicly available benchmarking data and comparative analysis of current methods provide a foundation for robust discovery of SV mosaicism in non-cancer tissues..
Conflict of interest statement
FJS receives research support from Illumina, PacBio and Oxford Nanopore. All other authors declare no conflict.
Figures




References
-
- Biesecker L.G., and Spinner N.B. (2013). A genomic view of mosaicism and human disease. Nat. Rev. Genet. 14, 307–320. - PubMed
-
- Campbell I.M., Yuan B., Robberecht C., Pfundt R., Szafranski P., McEntagart M.E., Nagamani S.C.S., Erez A., Bartnik M., Wiśniowiecka-Kowalnik B., et al. (2014). Parental somatic mosaicism is underrecognized and influences recurrence risk of genomic disorders. Am. J. Hum. Genet. 95, 173–182. - PMC - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources