

Spatially-restricted inflammation-induced senescent-like glia in multiple sclerosis and patient-derived organoids
- PMID: 41006208
- PMCID: PMC12475462
- DOI: 10.1038/s41467-025-63371-9
Spatially-restricted inflammation-induced senescent-like glia in multiple sclerosis and patient-derived organoids
Abstract
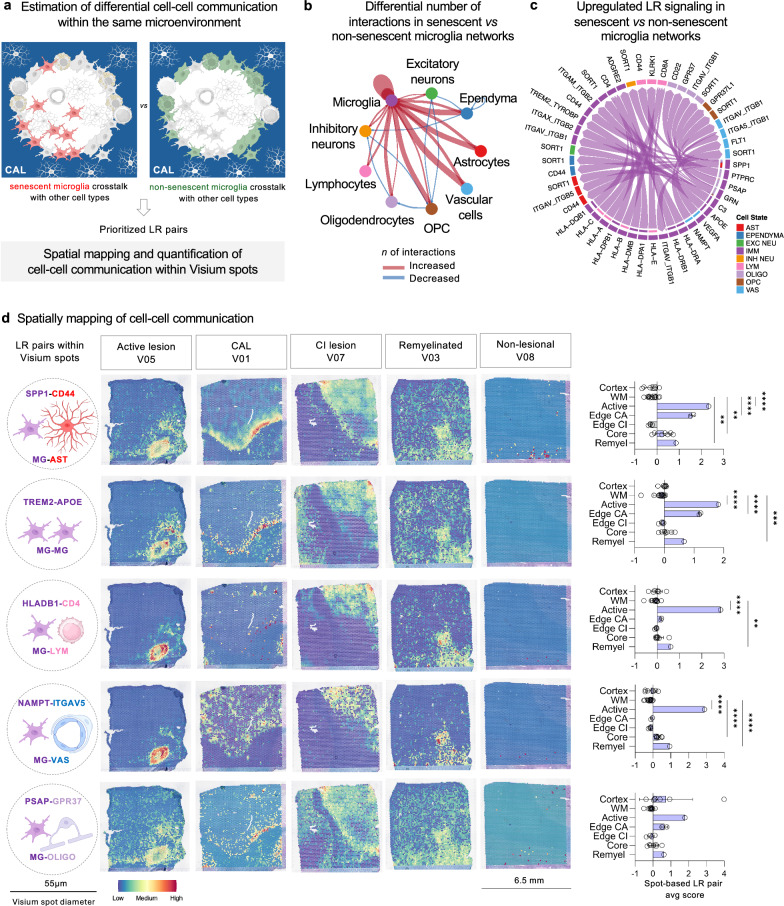
In multiple sclerosis (MS), chronic compartmentalized inflammation is thought to drive relentless clinical deterioration. Here, we investigate the link between unresolved parenchymal inflammation and cellular senescence in MS progression. Single-cell transcriptomic analysis of human brain tissue reveals an accumulation of senescent-like glial cells in diseased white matter, especially in chronic active lesions, and to a lesser extent in the cortex. Spatial transcriptomics show gradients of senescence-like signatures extending from lesion cores to periplaque regions, alongside rewired cellular networks. Experimental induction of senescence in MS hiPSC-derived neural organoids demonstrates that microglia are especially vulnerable to inflammation-induced senescence, which can be partially rescued by CNS-penetrant anti-inflammatory drugs. At the patient level (n = 466), increased 3T MRI-estimated brain-age is observed, especially in individuals with more than four chronic active lesions. These findings suggest that chronic inflammation might accelerate senescence-like processes, potentially contributing to disease progression, and that its modulation might help limit further propagation.
© 2025. The Author(s).
Conflict of interest statement
Competing interests: P.A.C. received research funding from Genentech and consulting honoraria for serving on advisory boards for Idorsia Spolia Therapeutics and Lilly. P.M. received consulting and/or speaker honoraria from Sanofi, Biogen, and Merck. D.S.R. received research funding from Abata Therapeutics and Sanofi. M.A. received consulting and/or speaker honoraria from GSK, Sanofi, Biogen, Immunic Therapeutics, Viatris, and Abata Therapeutics. All other authors report no relevant declaration of interest.
Figures









References
MeSH terms
Substances
Grants and funding
- 2022/R-Single/026/Fondazione Italiana Sclerosi Multipla (Italian Multiple Sclerosis Foundation)
- 2019-1677/Fondazione Cariplo (Cariplo Foundation)
- R01 NS082347/NS/NINDS NIH HHS/United States
- Intramural funding/U.S. Department of Health & Human Services | NIH | National Institute of Neurological Disorders and Stroke (NINDS)
- RFA-2412-44156/National Multiple Sclerosis Society (National MS Society)
LinkOut - more resources
Full Text Sources
Medical
