

SoMaCX: a complex generative genome modeling framework
- PMID: 41023610
- PMCID: PMC12482561
- DOI: 10.1186/s12864-025-12023-9
SoMaCX: a complex generative genome modeling framework
Abstract
Background: Somatic structural variations (SVs) are commonly observed in cancer tissue, but remain challenging to discover with short and long read sequencing due to tumor heterogeneity and other technical sequencing factors. Only SVs with a sufficient fraction of reads spanning the event will be detectable, while issues like chromothripsis increase the complexity and resulting interpretation significantly. Because structural variation is difficult to measure and reproduce in vivo, it is logical to make use of simulation frameworks to determine realistic system limitations. Our generative modeling approach called soMaCX uses distributions from data to empower simulations that approach real data.
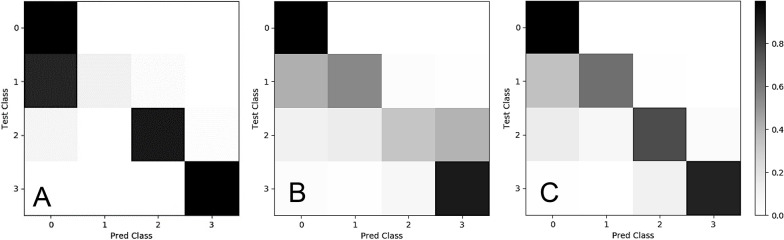
Results: Our generative framework includes mechanisms for biological conservation in the germline as well as tissue composition in the somatic along with regional distribution controls and complex SV generation that is not available in other systems. The output of this system is FASTA format which can then be used as input to any downstream read simulator making Illumina, PacBio, 10X genomics, Oxford-Nanopore and Bionano FASTQ data files which are further processed to become standard BAM files for SV calling.
Conclusions: The soMaCX framework provides superior generative modeling-based performance when compared to other simulation frameworks with respect to real data. Our open-source method introduces an important conceptual element to simulation by utilizing biological relevant regions (genes and regulatory elements) as the distribution controls along with the biological modulation of known pathways (end-joining) leading to more detailed and realistic simulated genomes. By designing a generative method to explore the most difficult genomic conditions, we provide a means to measure germline variation calling performance and to calibrate the results for rare variants needed in the clinical setting. We provide a python 3 implementation at: https://github.com/timothyjamesbecker/somacx .
Keywords: FASTA generation.; Generative modeling; Genome simulation; Next generation sequencing; Somatic simulation.
© 2025. The Author(s).
Conflict of interest statement
Declarations. Ethical approval: Not applicable. Consent for publication: Not applicable. Competing interests: The authors declare no competing interests.
Figures

References
-
- The Cancer Genome Atlas Research Network., Weinstein, J., Collisson, E. et al. The Cancer Genome Atlas Pan-Cancer analysis project. Nat Genet45, 1113–1120 (2013). 10.1038/ng.2764. https://www.nature.com/articles/ng.2764#citeas. - PMC - PubMed
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
