

This is a preprint.
A Common PD-Risk GBA1 Variant Disrupts LIMP2 Interaction, Impairs Glucocerebrosidase Function, and Drives Lysosomal and Mitochondrial Dysfunction
- PMID: 41030976
- PMCID: PMC12478413
- DOI: 10.1101/2025.08.28.672891
A Common PD-Risk GBA1 Variant Disrupts LIMP2 Interaction, Impairs Glucocerebrosidase Function, and Drives Lysosomal and Mitochondrial Dysfunction
Abstract
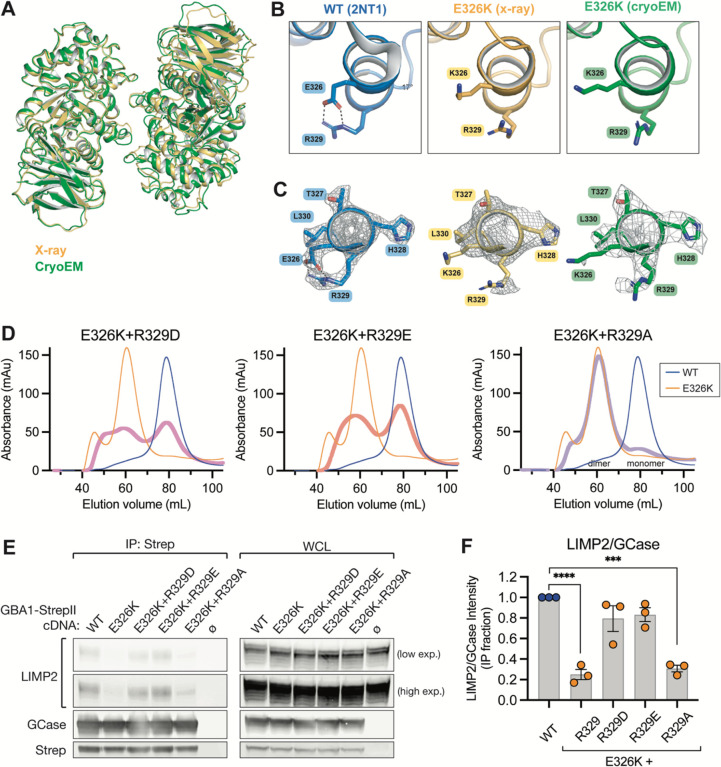
Variants in GBA1 cause Gaucher disease (GD), a lysosomal storage disorder, and represent the most common genetic risk factor for Parkinson's disease (PD). While some GBA1 variants are associated with both GD and PD, several coding mutations, including E326K, specifically confer risk for developing PD. It is established that GD-linked variants in β-glucocerebrosidase (GCase), the enzyme encoded by GBA1, are loss-of-function, but it remains unclear whether variants solely associated with PD similarly reduce GCase activity. The mechanisms by which some of these variants impact GCase activity and PD-associated pathways, including lysosomal and mitochondrial function, are also poorly defined. Here, we show that the PD-linked E326K variant significantly reduces lysosomal GCase activity by impairing its delivery to lysosomes via altered interactions with its receptor, LIMP2. Biophysical and structural characterization of this variant, both alone and in complex with LIMP2, reveals a dimeric organization that appears to result from the loss of a key salt bridge between E326 and R329. Restoration of this salt bridge through the introduction of a negatively charged side chain at position 329 promotes monomeric organization and interaction with LIMP2 in cells. GBA1-p.E326K cell models show greater deficits in PD-linked pathways compared to more severe loss of GCase function, including secondary lysosomal lipid storage and mitochondrial dysfunction. We confirm the E326K variant impacts GCase pathway activity in relevant CNS cell types, including iPSC-derived microglia, and in biofluids from heterozygous GBA1-p.E326K variant carriers. Together, our data provide key insights into the nature of GCase dysfunction in GBA1-PD and can inform the development of GCase-targeted therapeutic strategies to treat PD.
Conflict of interest statement
Competing interests The authors declare the following competing interests: O.B.D, L.R., J.E.K., S.S.D., R.G., S.V.A., N.S.G., J.H.K., E.T., M.A., J.P.C., M.T.M., A.H.N., H.N.N., N.E.P., E.I.L., T.Y., K.X., R.A.P.S., R.L., J.C.U., S.B., H.P.B., M.P., A.N.Q., X.W., G.D.P., M.S.K., C.S.M., A.A., J.H.S., and A.G.H. were full time employees and shareholders of Denali Therapeutics during the course of this work. X.W. and M.A. are currently employees of Tenvie Therapeutics.
Figures







References
-
- Hertz E., Chen Y., and Sidransky E. (2024). Gaucher disease provides a unique window into Parkinson disease pathogenesis. Preprint at Nature Research, https://doi.org/10.1038/s41582-024-00999-z 10.1038/s41582-024-00999-z. - DOI
Publication types
LinkOut - more resources
Full Text Sources
Miscellaneous