

Tracking down the origin and subsequent spread of SARS-CoV-2 lineage B.1.619
- PMID: 41031290
- PMCID: PMC12477603
- DOI: 10.1093/ve/veaf017
Tracking down the origin and subsequent spread of SARS-CoV-2 lineage B.1.619
Abstract
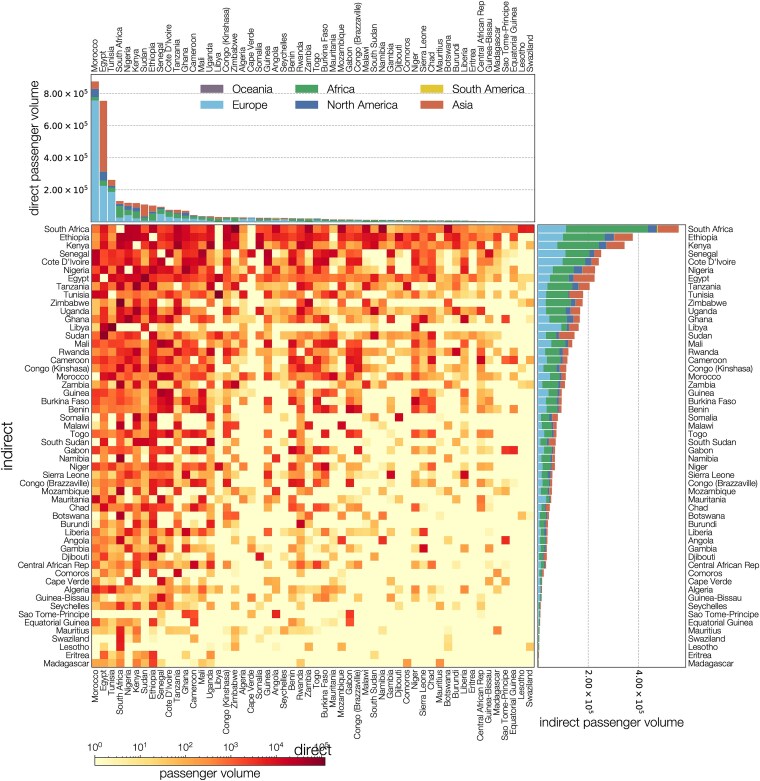
Since late 2020, the emergence of variants of concern (VOCs) of SARS-CoV-2 has been of concern to public health, researchers and policymakers. Mutations in the SARS-CoV-2 genome-for which clear evidence is available indicating a significant impact on transmissibility, severity and/or immunity-illustrate the importance of genomic surveillance and monitoring the evolution and geographic spread of novel lineages. Lineage B.1.619 was first detected in Switzerland in January 2021, in international travellers returning from Cameroon. This lineage was subsequently also detected in Rwanda, Belgium, Cameroon, France, and many other countries and is characterised by spike protein amino acid mutations N440K and E484K in the receptor binding domain, which are associated with immune escape and higher infectiousness. In this study, we perform a phylogeographic analysis to track the geographic origin and subsequent dispersal of SARS-CoV-2 lineage B.1.619. We employ a recently developed travel history-aware phylogeographic model, enabling us to incorporate genomic sequences with associated travel information. We estimate that B.1.619 most likely originated in Cameroon, in November 2020. We estimate the influence of the number of air-traffic passengers on the dispersal of B.1.619 but find no significant effect, illustrative of the complex dispersal patterns of SARS-CoV-2 lineages. Finally, we examine the metadata associated with infected Belgian patients and report a wide range of symptoms and medical interventions.
Keywords: B.1.619; Bayesian inference; COVID-19; GLM; Markov chain Monte Carlo; SARS-CoV-2; air traffic; phylogenetics; phylogeography.
© The Author(s) 2025. Published by Oxford University Press.
Figures







References
LinkOut - more resources
Full Text Sources
Miscellaneous
