

This is a preprint.
Targeted glycophagy ATG8 therapy reverses diabetic heart disease in mice and in human engineered cardiac tissues
- PMID: 41040214
- PMCID: PMC12485856
- DOI: 10.1101/2024.11.28.625926
Targeted glycophagy ATG8 therapy reverses diabetic heart disease in mice and in human engineered cardiac tissues
Update in
-
Targeted glycophagy ATG8 therapy reverses diabetic heart disease in mice and in human engineered cardiac tissues.Nat Cardiovasc Res. 2025 Nov;4(11):1487-1500. doi: 10.1038/s44161-025-00726-x. Epub 2025 Sep 29. Nat Cardiovasc Res. 2025. PMID: 41023280
Abstract
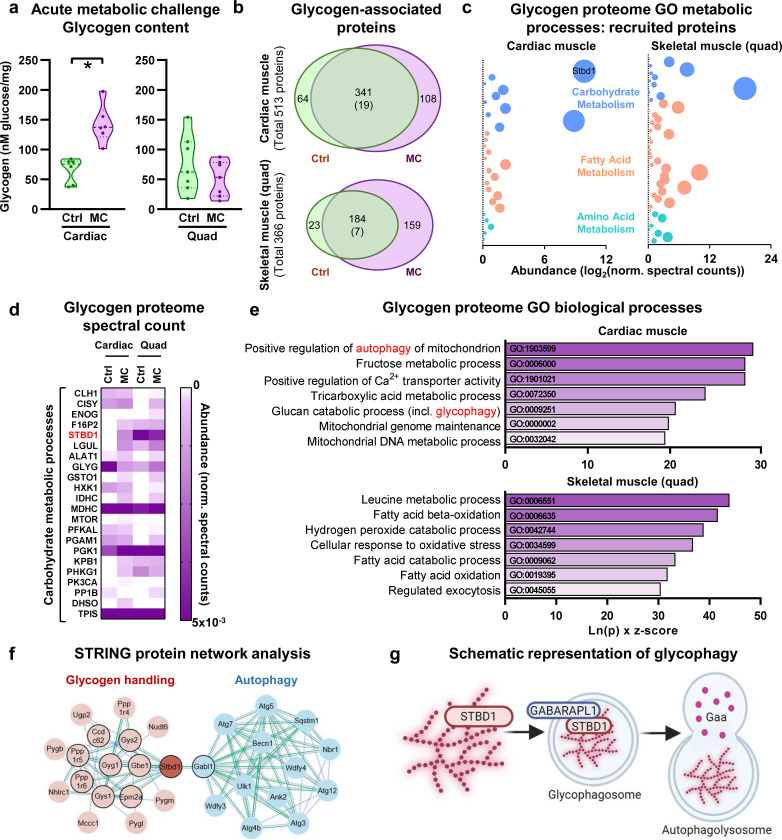
Diabetic heart disease is highly prevalent and is associated with the early development of impaired diastolic relaxation. The mechanisms of diabetic heart disease are poorly understood and it is a condition for which there are no targeted therapies. Recently, disrupted glycogen-autophagy (glycophagy) and glycogen accumulation have been identified in the diabetic heart. Glycophagy involves glycogen receptor binding and linking with an ATG8 protein to locate and degrade glycogen within an intracellular phago-lysosome. Here we show that glycogen receptor protein STBD1 (starch-binding-domain-protein-1) is mobilized early in the cardiac glycogen response to metabolic challenge in vivo, and that deficiency of a specific ATG8 family protein, Gabarapl1 (γ-aminobutyric-acid-receptor-associated-protein-like-1) is associated with diastolic dysfunction in diabetes. Gabarapl1 gene delivery treatment remediated cardiomyocyte and cardiac diastolic dysfunction in type 2 diabetic mice and diastolic performance of 'diabetic' human iPSC-derived cardiac organoids. We identify glycophagy dysregulation as a mechanism and potential treatment target for diabetic heart disease.
Conflict of interest statement
COMPETING INTERESTS The authors declare no competing financial interests.
Figures






References
-
- Mellor K. M. et al. Myocardial glycophagy flux dysregulation and glycogen accumulation characterize diabetic cardiomyopathy. Journal of Molecular and Cellular Cardiology 189, 83–89 (2024). - PubMed
-
- Koutsifeli P. et al. Glycogen-autophagy: Molecular machinery and cellular mechanisms of glycophagy. J Biol Chem 298, 102093 (2022).
-
- Varma U., Koutsifeli P., Benson V. L., Mellor K. M. & Delbridge L. M. D. Molecular mechanisms of cardiac pathology in diabetes - Experimental insights. Biochim Biophys Acta Mol Basis Dis 1864, 1949–1959 (2018). - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials