

Superinfection promotes replication and diversification of defective HIV-1 proviruses in people with non-suppressible viraemia
- PMID: 41044362
- PMCID: PMC12578631
- DOI: 10.1038/s41564-025-02135-z
Superinfection promotes replication and diversification of defective HIV-1 proviruses in people with non-suppressible viraemia
Abstract
During replication of some RNA viruses, defective particles can spontaneously arise and interfere with wild-type (WT) virus replication. However, these defective interfering particles (DIPs) have not been reported in people with HIV-1 (PWH). Here we find DIPs in PWH who have a rare, polyclonal form of non-suppressible viraemia (NSV). We characterized the source of NSV in two PWH who never reached undetectable viral load despite adherence to antiretroviral therapy (ART). Remarkably, in each participant, we found a diverse set of defective viral genomes sharing the same fatal deletions. This paradoxical accumulation of mutations by viruses with fatal defects was driven by superinfection with intact viruses, resulting in mobilization of defective genomes and accumulation of additional mutations during untreated infection. These defective proviruses interfere with WT virus replication, conditionally replicate and, in one case, have an R0 > 1, enabling in vivo spread. Despite this, clinical outcomes showed no beneficial effect of these DIPs. These findings demonstrate that fatally defective proviruses, traditionally considered evolutionary dead ends, can replicate and diversify upon superinfection without preventing disease progression.
© 2025. The Author(s).
Conflict of interest statement
Competing interests: R.F.S. is an inventor on a patent application for the intact proviral DNA assay (IPDA) (patent no. PCT/US16/28822) filed by Johns Hopkins University and licensed by AccelevirDx. F.R.S. received payments from Gilead Sciences for participating at scientific meetings. The other authors declare no competing interests.
Figures



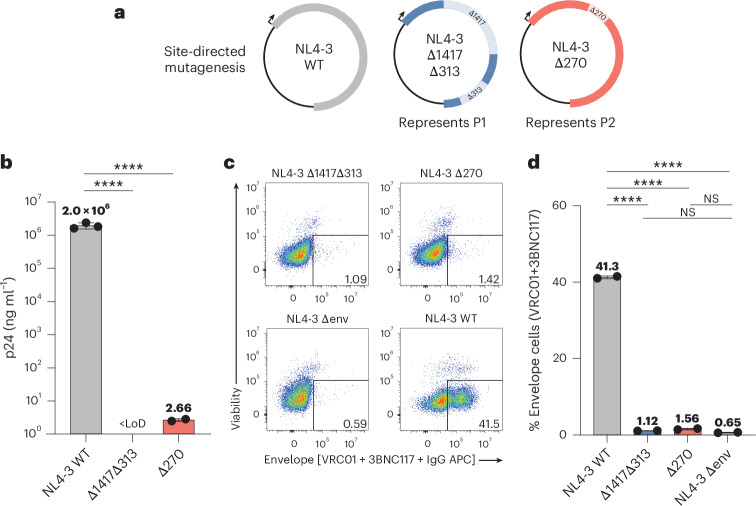
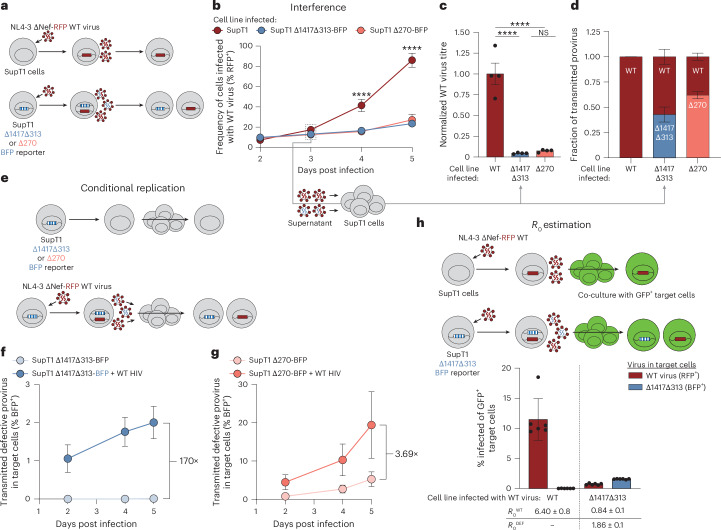







Update of
-
Superinfection with intact HIV-1 results in conditional replication of defective proviruses and nonsuppressible viremia in people living with HIV-1.bioRxiv [Preprint]. 2025 Apr 4:2025.04.04.647291. doi: 10.1101/2025.04.04.647291. bioRxiv. 2025. Update in: Nat Microbiol. 2025 Nov;10(11):2736-2748. doi: 10.1038/s41564-025-02135-z. PMID: 40236094 Free PMC article. Updated. Preprint.
References
-
- Chun, T.-W. et al. In vivo fate of HIV-1-infected T cells: quantitative analysis of the transition to stable latency. Nat. Med.1, 1284–1290 (1995). - PubMed
-
- Chun, T.-W. et al. Quantification of latent tissue reservoirs and total body viral load in HIV-1 infection. Nature387, 183–188 (1997). - PubMed
-
- Finzi, D. et al. Identification of a reservoir for HIV-1 in patients on highly active antiretroviral therapy. Science278, 1295–1300 (1997). - PubMed
-
- Wong, J. K. et al. Recovery of replication-competent HIV despite prolonged suppression of plasma viremia. Science278, 1291–1295 (1997). - PubMed
MeSH terms
Substances
Grants and funding
- U01DA036935/U.S. Department of Health & Human Services | NIH | National Institute on Drug Abuse (NIDA)
- P30AI094189/Johns Hopkins | School of Medicine, Johns Hopkins University (SOM, JHU)
- P30 AI094189/AI/NIAID NIH HHS/United States
- U01 DA036935/DA/NIDA NIH HHS/United States
- DP5OD031834/U.S. Department of Health & Human Services | NIH | National Institute of Dental and Craniofacial Research (NIDCR)
LinkOut - more resources
Full Text Sources
Medical
