

Ferroptosis-induced SUMO2 lactylation counteracts ferroptosis by enhancing ACSL4 degradation in lung adenocarcinoma
- PMID: 41057295
- PMCID: PMC12504568
- DOI: 10.1038/s41421-025-00829-6
Ferroptosis-induced SUMO2 lactylation counteracts ferroptosis by enhancing ACSL4 degradation in lung adenocarcinoma
Abstract
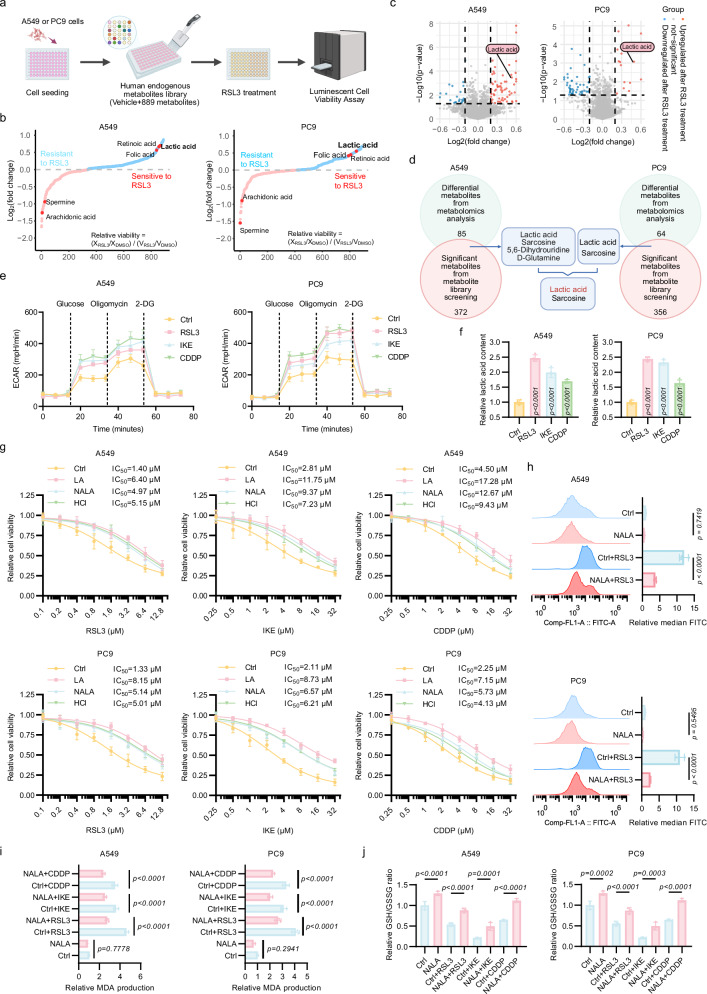
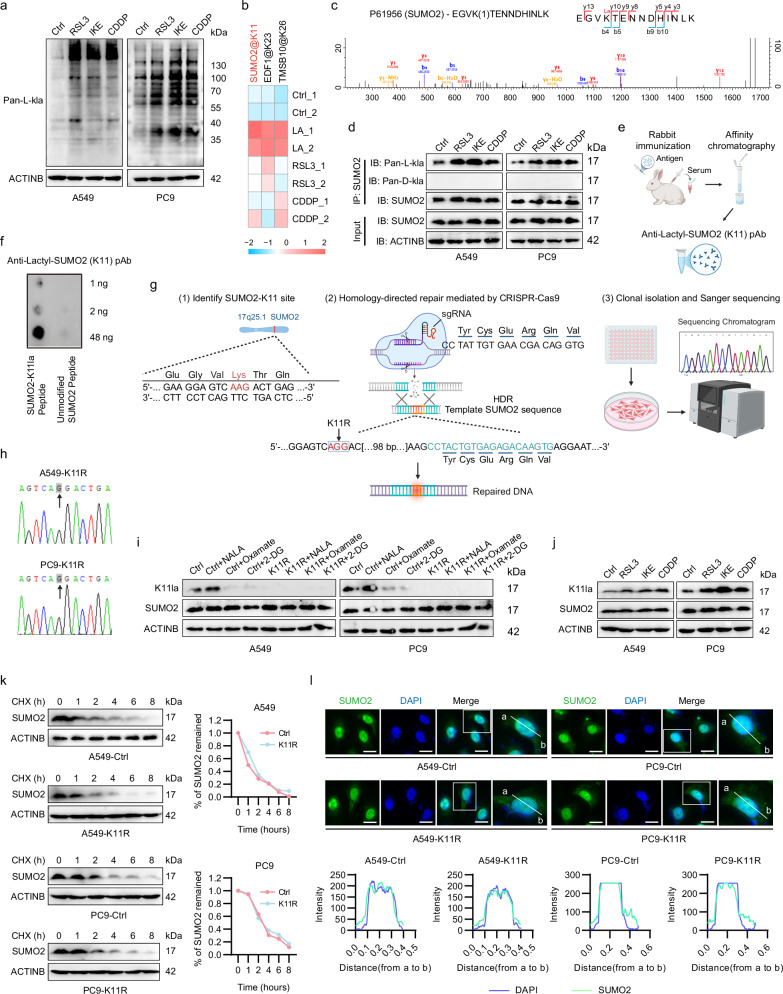
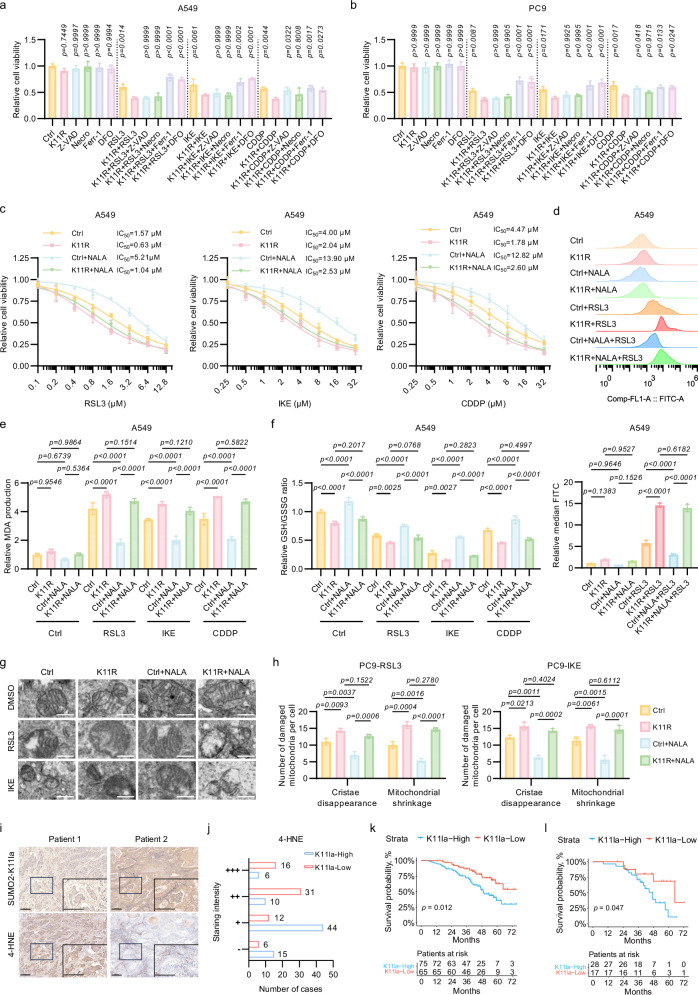
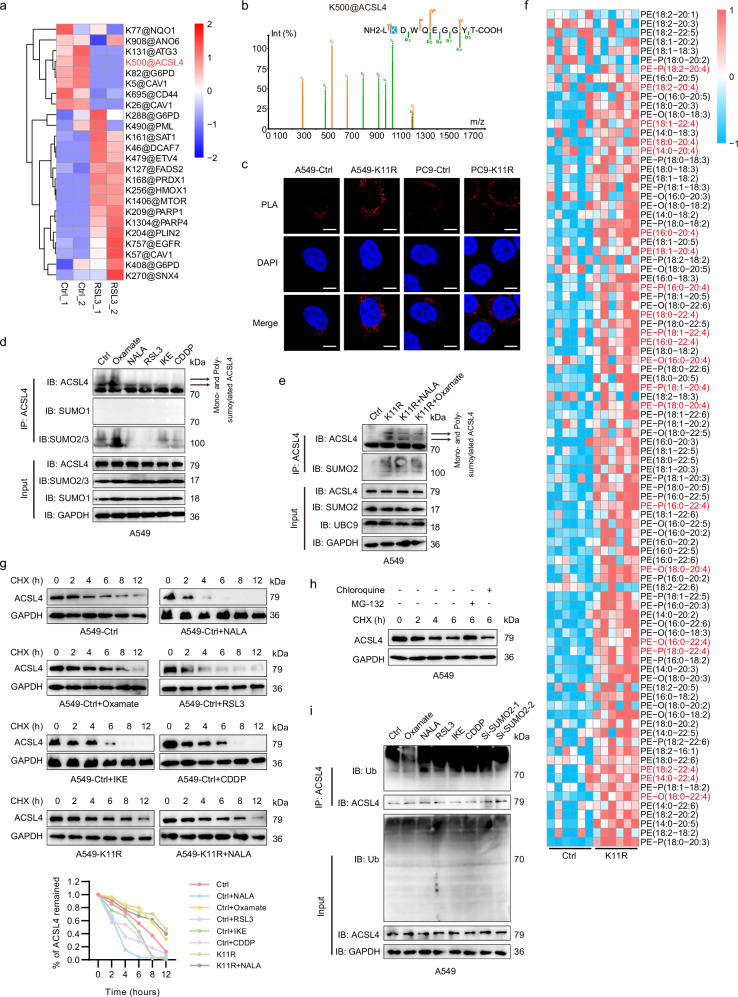
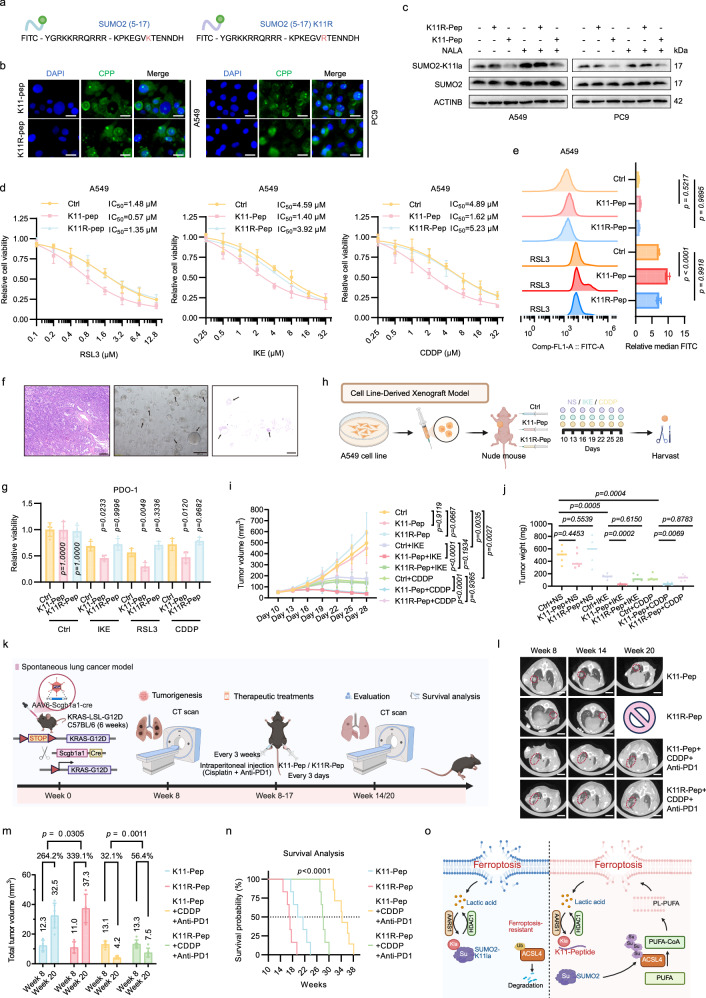
Lactylation, a lactate-mediated post-translational modification, has garnered significant attention for its pivotal role in epigenetic modulation. However, the intricate interplay between lactylation and ferroptosis in lung adenocarcinoma (LUAD) remains to be fully elucidated. Utilizing metabolomic profiling and comprehensive metabolic library screening, our study uncovers that ferroptosis markedly enhances lactic acid accumulation and subsequent protein lactylation, which in turn confers resistance to ferroptosis in LUAD cells. Functional assays, comprising cell viability tests, lipid peroxidation detection, as well as malondialdehyde and glutathione measurements, collectively reveal that SUMO2-K11 lactylation (SUMO2-K11la), the most prominently elevated lactylation in response to ferroptosis induction, serves as a pivotal factor in determining ferroptosis resistance. Sumoylation proteomics and co-immunoprecipitation assays reveal that SUMO2-K11la impairs the interaction between SUMO2 and ACSL4. Consequently, this disruption facilitates the degradation of ACSL4, thereby disrupting lipid metabolism and effectively mitigating ferroptosis. Furthermore, AARS1 is identified as the lactyltransferase and HDAC1 as the delactylase for SUMO2-K11la. Based on these findings, we develop a cell-penetrating peptide that competitively and specifically inhibits SUMO2-K11la. This peptide significantly potentiates ferroptosis and sensitizes LUAD to cisplatin in xenograft models, while enhancing chemoimmunotherapy responses in spontaneous lung cancer models. Overall, our findings imply that SUMO2-K11la is a pivotal regulator of ferroptosis resistance in LUAD, and suggest a promising strategy to potentiate ferroptosis-based cancer therapies via targeting SUMO2-K11la by the cell-penetrating peptide.
© 2025. The Author(s).
Conflict of interest statement
Competing interests: The authors declare no competing interests. Ethical approval: All participants have signed the informed consent. This study adhered to the Declaration of Helsinki and was approved by the Zhongshan Hospital Research Ethics Committee (B2022-180R).
Figures


References
-
- Bray, F. et al. Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin.74, 229–263 (2024). - PubMed
-
- Gridelli, C. et al. Non-small-cell lung cancer. Nat. Rev. Dis. Prim.1, 15009 (2015). - PubMed
-
- Miller, K. D. et al. Cancer treatment and survivorship statistics, 2022. CA Cancer J. Clin.72, 409–436 (2022). - PubMed
-
- Hanahan, D. Hallmarks of cancer: new dimensions. Cancer Discov.12, 31–46 (2022). - PubMed
-
- Hanahan, D. & Weinberg, R. A. Hallmarks of cancer: the next generation. Cell144, 646–674 (2011). - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
