

Scalable long-read nanopore HPV16 amplicon-based whole-genome sequencing
- PMID: 41057388
- PMCID: PMC12504524
- DOI: 10.1038/s41598-025-18664-w
Scalable long-read nanopore HPV16 amplicon-based whole-genome sequencing
Abstract
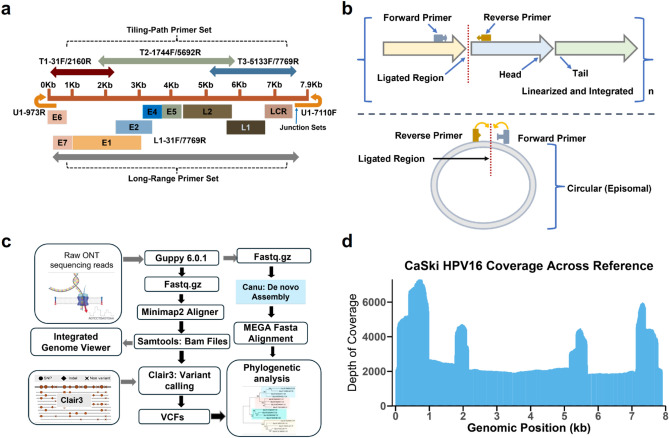
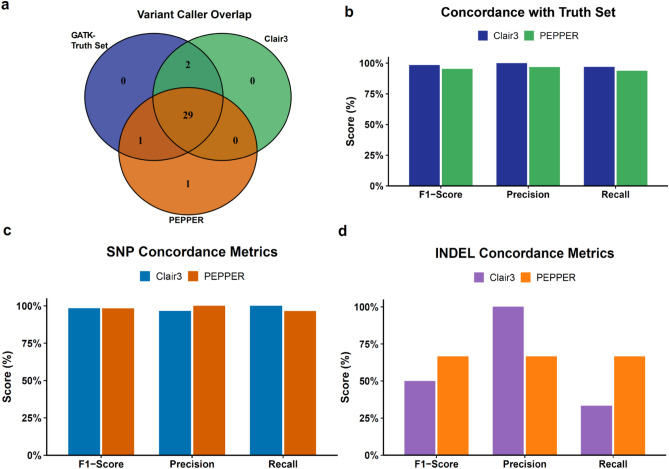
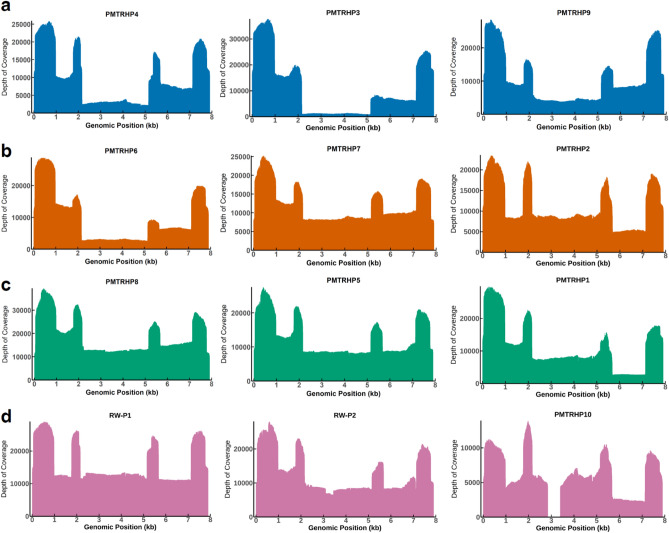
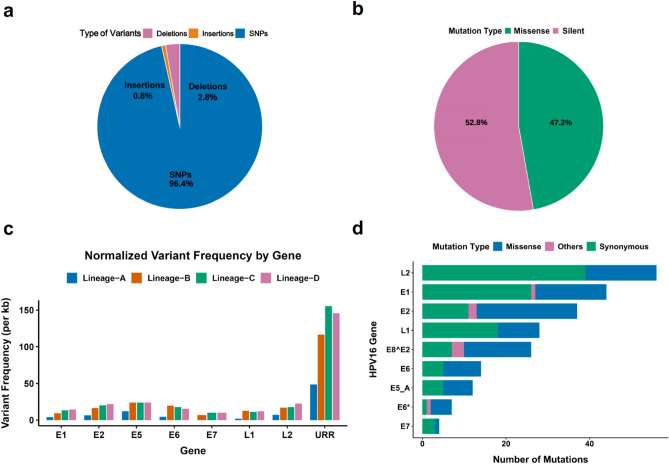
Human papillomavirus 16 (HPV16) drives precursor cervical lesions that often progress to cervical cancer (CC). Variation within the HPV16 genome has been associated with CC risk. Here, we developed an affordable and portable amplicon-based long-read whole genome sequencing (WGS) approach using Oxford Nanopore Technologies to investigate HPV16 genetic diversity among women in sub-Saharan African countries. Applied to a control CaSki cell line and clinical samples (n = 12), our method generated complete HPV16 genomes at high coverage (median read coverage: 5,899-15,279 ×). Benchmarking our HPV16 controls showed high accuracy for two variant calling pipelines (Clair3 and PEPPER-Margin DeepVariant). Phylogenetic analysis identified all four previously defined HPV16 lineages (A-D) and their high-risk sublineages. All lineages exhibited strong concordance across de novo assembly, reference-based phylogenetics, and unsupervised clustering. Our pipeline effectively captured the full extent of genomic variation, including putative lineage-informative SNPs. This method offers a robust amplicon-based WGS and analysis pipeline for HPV16, making it well-suited for integration into surveillance, diagnostics, and epidemiological efforts in low-resource areas.
© 2025. The Author(s).
Conflict of interest statement
Declarations. Competing interests: The authors declare no competing interests. Ethical statement: All participants provided written informed consent. Ethical approvals were obtained from the Institutional Research and Ethics Committee (IREC) of Moi Teaching and Referral Hospital (MTRH) and Moi University, Eldoret, Kenya (reference number IREC/371/2022) and from the Institutional Review Board (IRB) of Brown University, Providence, Rhode Island, USA (IRB-ID: 2,023,003,553). All study procedures were performed in accordance with relevant guidelines and regulations outlined by the Ethics Review Boards indicated above.
Figures

Update of
-
Scalable long-read Nanopore HPV16 Amplicon-based Whole-Genome Sequencing.medRxiv [Preprint]. 2025 May 23:2025.05.22.25328149. doi: 10.1101/2025.05.22.25328149. medRxiv. 2025. Update in: Sci Rep. 2025 Oct 7;15(1):34892. doi: 10.1038/s41598-025-18664-w. PMID: 40661276 Free PMC article. Updated. Preprint.
References
-
- Sung, H. et al. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA: A Cancer Journal for Clinicians vol. 71 209–249 Preprint at 10.3322/caac.21660 (2021). - PubMed
-
- Sengayi-Muchengeti, M. et al. Cervical cancer survival in sub-Saharan Africa by age, stage at diagnosis and Human Development Index: A population-based registry study. Int. J. Cancer147, 3037–3048 (2020). - PubMed
-
- World Health Organization. Global Strategy to Accelerate the Elimination of Cervical Cancer as a Public Health Problem.
-
- Recommendations and Good Practice Statements on Screening and Treatment to Prevent Cervical Cancer. (World Health Organization, 2021).
MeSH terms
Grants and funding
- P30AI042853/Providence/Boston CFAR, United States of America
- U54 CA254518/CA/NCI NIH HHS/United States
- D43TW011317/National Institutes of Health (NIH), United States of America
- U54 CA190151/CA/NCI NIH HHS/United States
- 5U54CA190151-02 and 5U54CA254518-03/National Institutes of Health (NIH), National Cancer Institute (NCI), United States of America
LinkOut - more resources
Full Text Sources
