

Genotyping short tandem repeats across copy number alterations, aneuploidies, and polyploid organisms
- PMID: 41057506
- PMCID: PMC12504596
- DOI: 10.1038/s42003-025-08837-8
Genotyping short tandem repeats across copy number alterations, aneuploidies, and polyploid organisms
Abstract
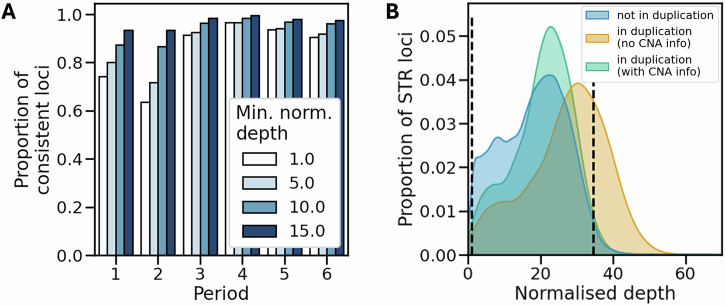
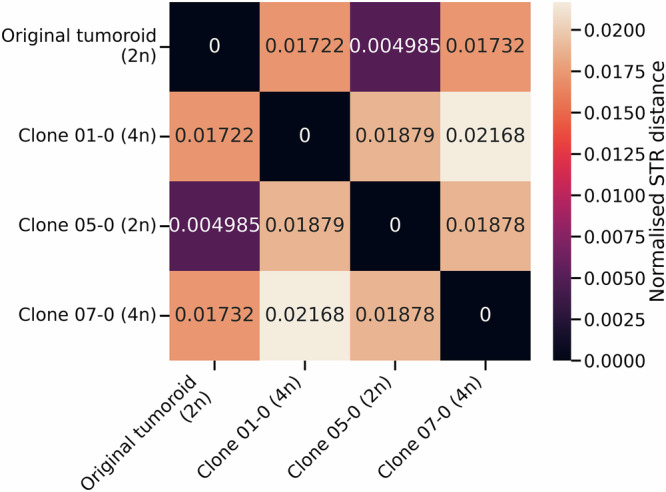
Short tandem repeats (STRs) are a rich source of genetic variation, but are difficult to genotype. While specialized repeat variant callers exist, they typically assume a euploid human genome. This means recent findings regarding phenotypic effects of STR variants in human health and disease cannot be readily extended to polyploid organisms or cancer, which is characterised by copy number alterations (CNAs). Here we present ConSTRain, a novel STR variant caller that explicitly accounts for the copy number of loci in its genotyping approach. We benchmark ConSTRain using a euploid human 100X whole genome sequencing sample where it calls STR allele lengths for over 1.7 × 106 loci in under 20 minutes with an accuracy of 98.28%. Subsequently, we show that ConSTRain resolves complex STR genotypes in an artificial trisomy 21 sample and a polyploid Dwarf Cavendish banana harbouring a large duplication. Finally, we analyse a microsatellite instable colorectal cancer tumoroid, where ConSTRain tackles CNAs and whole-genome duplications. ConSTRain is the first STR variant caller that allows for the investigation of repeats affected by CNAs, aneuploidies, and polyploid genomes. This unlocks the investigation of STRs across a wide range of contexts and organisms where they previously could not be easily studied.
© 2025. The Author(s).
Conflict of interest statement
Competing interests: M.A. is an Editorial Board Member for Communications Biology, but was not involved in the editorial review of, nor the decision to publish this article. The other authors declare no competing interests.
Figures


References
MeSH terms
LinkOut - more resources
Full Text Sources
