

Sperm sequencing reveals extensive positive selection in the male germline
- PMID: 41062690
- PMCID: PMC12611766
- DOI: 10.1038/s41586-025-09448-3
Sperm sequencing reveals extensive positive selection in the male germline
Abstract
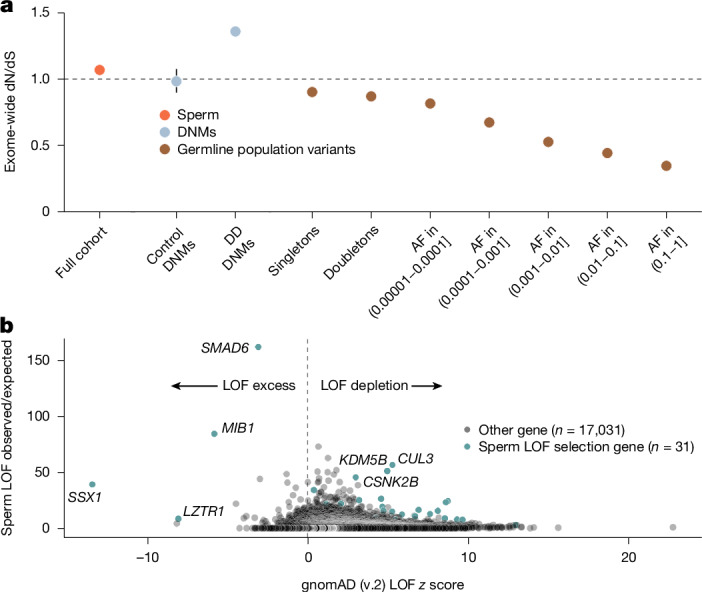
Mutations that occur in the cell lineages of sperm or eggs can be transmitted to offspring. In humans, positive selection of driver mutations during spermatogenesis can increase the birth prevalence of certain developmental disorders1-3. Until recently, characterizing the extent of this selection in sperm has been limited by the error rates of sequencing technologies. Here we used the duplex sequencing method NanoSeq4 to sequence 81 bulk sperm samples from individuals aged 24-75 years. Our findings revealed a linear accumulation of 1.67 (95% confidence interval of 1.41-1.92) mutations per year per haploid genome driven by two mutational signatures associated with human ageing. Deep targeted and exome NanoSeq5 of sperm samples identified more than 35,000 germline coding mutations. We detected 40 genes (31 newly identified) under significant positive selection in the male germline that have activating or loss-of-function mechanisms and are involved in diverse cellular pathways. Most of the positively selected genes are associated with developmental or cancer predisposition disorders in children, whereas four of the genes exhibited increased frequencies of protein-truncating variants in healthy populations. We show that positive selection during spermatogenesis drives a 2-3-fold increased risk of known disease-causing mutations, which results in 3-5% of sperm from middle-aged to older individuals with a pathogenic mutation across the exome. These findings shed light on germline selection dynamics and highlight a broader increased disease risk for children born to fathers of advanced age than previously appreciated.
© 2025. Crown.
Conflict of interest statement
Competing interests: I.M., M.R.S. and P.J.C. are co-founders, shareholders and consultants for Quotient Therapeutics. R.E.A. is an employee of Quotient Therapeutics. M.E.H. is a co-founder of, consultant to and holds shares in Congenica, a genetics diagnostic company. The remaining authors declare no competing interests.
Figures












References
-
- Goriely, A., McVean, G. A. T., Röjmyr, M., Ingemarsson, B. & Wilkie, A. O. M. Evidence for selective advantage of pathogenic FGFR2 mutations in the male germ line. Science301, 643–646 (2003). - PubMed
-
- Abascal, F. et al. Somatic mutation landscapes at single-molecule resolution. Nature593, 405–410 (2021). - PubMed
-
- Lawson, A. R. J. et al. Somatic mutation and selection at population scale. Nature10.1038/s41586-025-09584-w (2025).
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
