

Sex and smoking bias in the selection of somatic mutations in human bladder
- PMID: 41062697
- PMCID: PMC12611770
- DOI: 10.1038/s41586-025-09521-x
Sex and smoking bias in the selection of somatic mutations in human bladder
Abstract
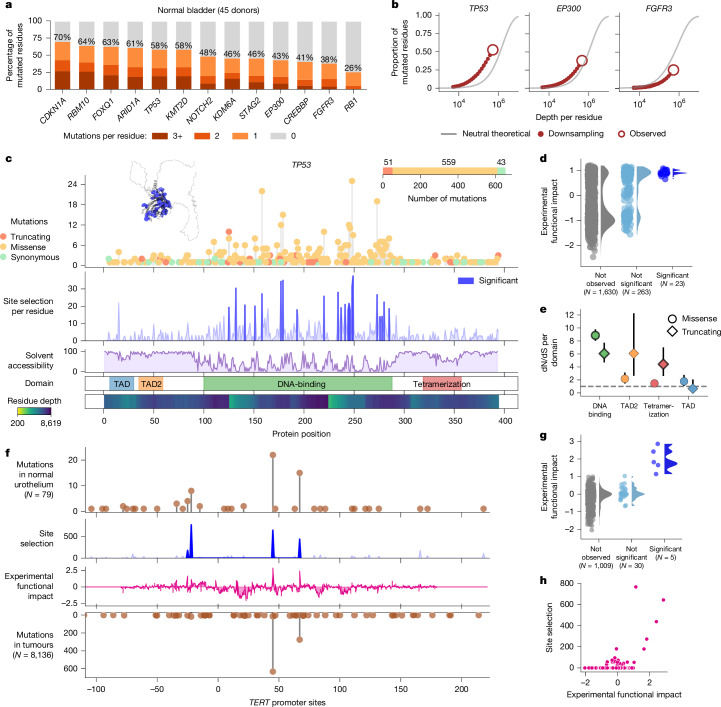
Men are at higher risk of several cancer types than women1. For bladder cancer the risk is four times higher for reasons that are not clear2. Smoking is also a principal risk factor for several tumour types, including bladder cancer3. As tumourigenesis is driven by somatic mutations, we wondered whether the landscape of clones in the normal bladder differs by sex and smoking history. Using ultradeep duplex DNA sequencing (approximately 5,000×), we identified thousands of clonal driver mutations in 16 genes across 79 normal bladder samples from 45 people. Men had significantly more truncating driver mutations in RBM10, CDKN1A and ARID1A than women, despite similar levels of non-protein-affecting mutations. This result indicates stronger positive selection on driver truncating mutations in these genes in the male urothelium. We also found activating TERT promoter mutations driving clonal expansions in the normal bladder that were associated strongly with age and smoking. These findings indicate that bladder cancer risk factors, such as sex and smoking, shape the clonal landscape of the normal urothelium. The high number of mutations identified by this approach offers a new strategy to study the functional effect of thousands of mutations in vivo-natural saturation mutagenesis-that can be extended to other human tissues.
© 2025. The Author(s).
Conflict of interest statement
Competing interests: R.A.R. is an equity holder at TwinStrand Biosciences Inc. and NanoString Technologies Inc. R.A.R. is named inventor on patent no. 11,479,807 (Methods for targeted nucleic acid sequence enrichment with applications to error corrected nucleic acid sequencing) owned by the University of Washington and licensed to TwinStrand Biosciences Inc. R.A.R. was a consultant at TwinStrand Biosciences Inc. and received research funding from a joint research grant with TwinStrand Biosciences Inc. and Ovartec GmbH. B.F.K. is an equity holder at NanoString Technologies Inc. The other authors declare no competing interests.
Figures














References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
