

Multi-omic network inference from time-series data
- PMID: 41087386
- PMCID: PMC12521560
- DOI: 10.1038/s41540-025-00591-1
Multi-omic network inference from time-series data
Abstract
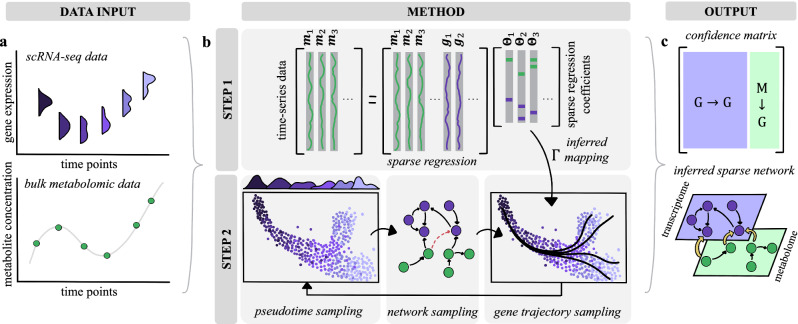
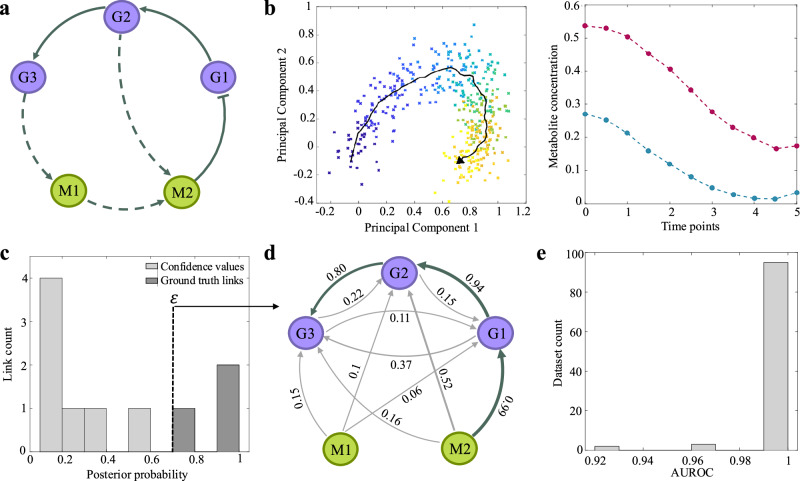
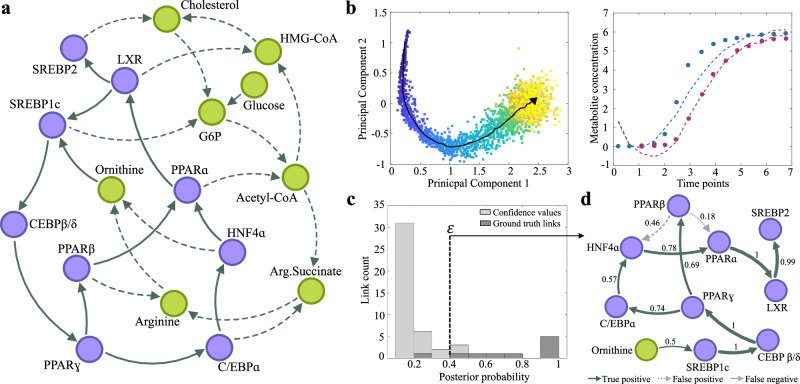
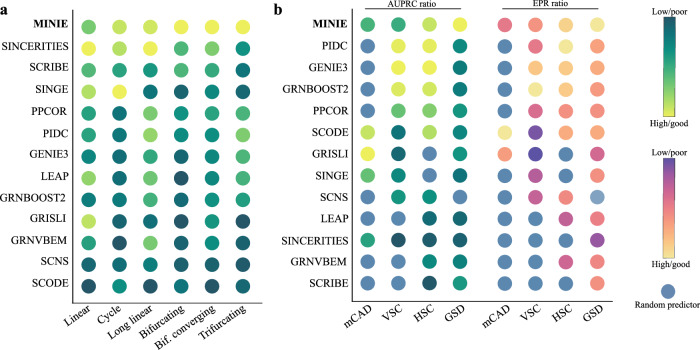
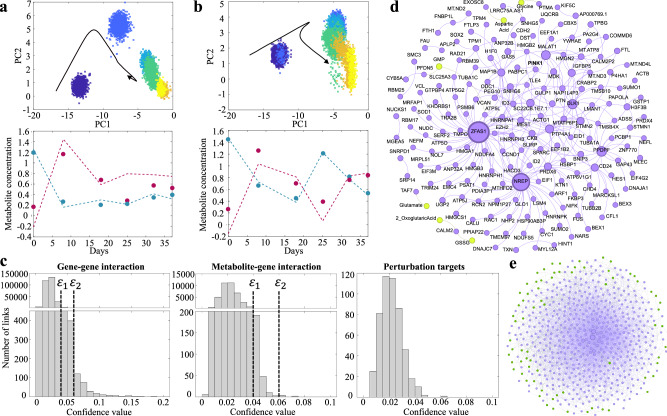
Biological phenotypes emerge from complex interactions across molecular layers. Yet, data-driven approaches to infer these regulatory networks have primarily focused on single-omic studies, overlooking inter-layer regulatory relationships. To address these limitations, we developed MINIE, a computational method that integrates multi-omic data from bulk metabolomics and single-cell transcriptomics through a Bayesian regression approach that explicitly models the timescale separation between molecular layers. We validate the method on both simulated datasets and experimental Parkinson's disease data. MINIE exhibits accurate and robust predictive performance across and within omic layers, including curated multi-omic networks and the lac operon. Benchmarking demonstrated significant improvements over state-of-the-art methods while ranking among the top performers in comprehensive single-cell network inference analysis. The integration of regulatory dynamics across molecular layers and temporal scales provides a powerful tool for comprehensive multi-omic network inference.
© 2025. The Author(s).
Conflict of interest statement
Competing interests: The authors declare no competing interests.
Figures


References
-
- Yasemi, M. & Jolicoeur, M. Modelling cell metabolism: a review on constraint-based steady-state and kinetic approaches. Processes9, 322 (2021).
MeSH terms
LinkOut - more resources
Full Text Sources
