

Decreased SNCA expression in whole-blood RNA analysis of Parkinson's disease adjusting for neutrophils
- PMID: 41087437
- PMCID: PMC12521519
- DOI: 10.1038/s41531-025-01062-4
Decreased SNCA expression in whole-blood RNA analysis of Parkinson's disease adjusting for neutrophils
Abstract
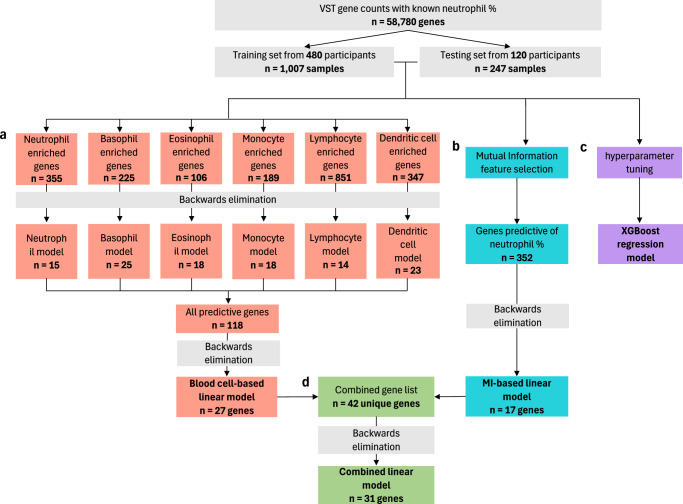
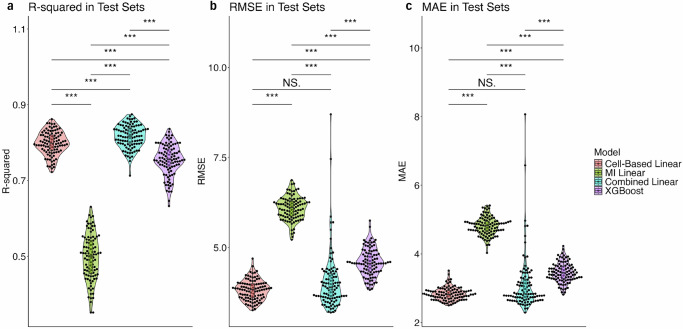
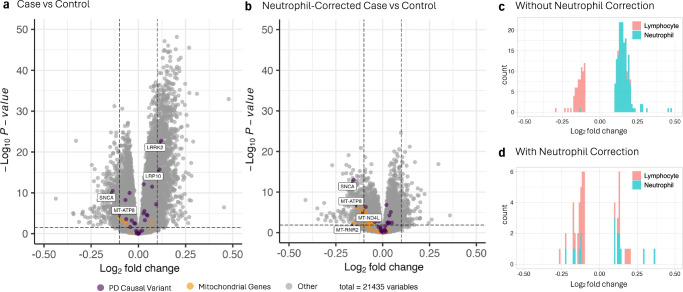
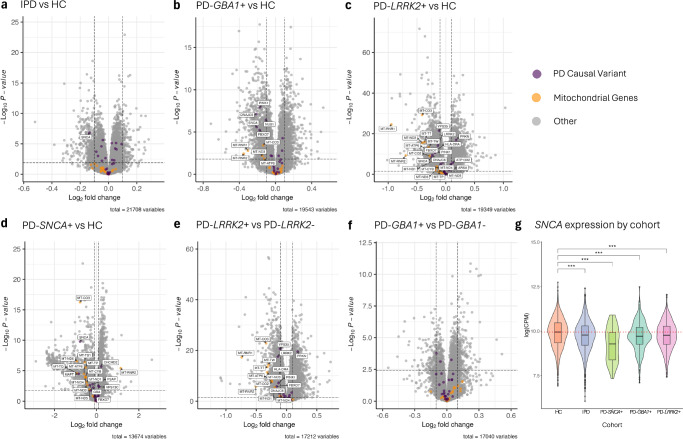
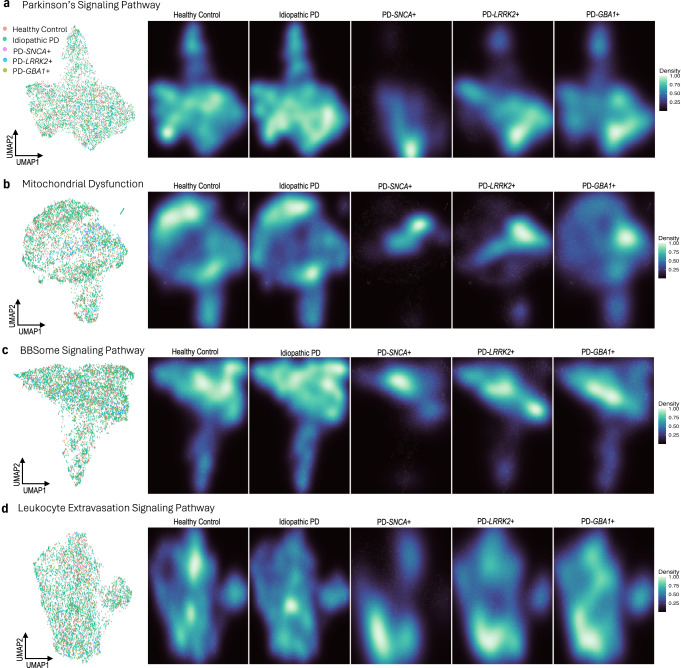
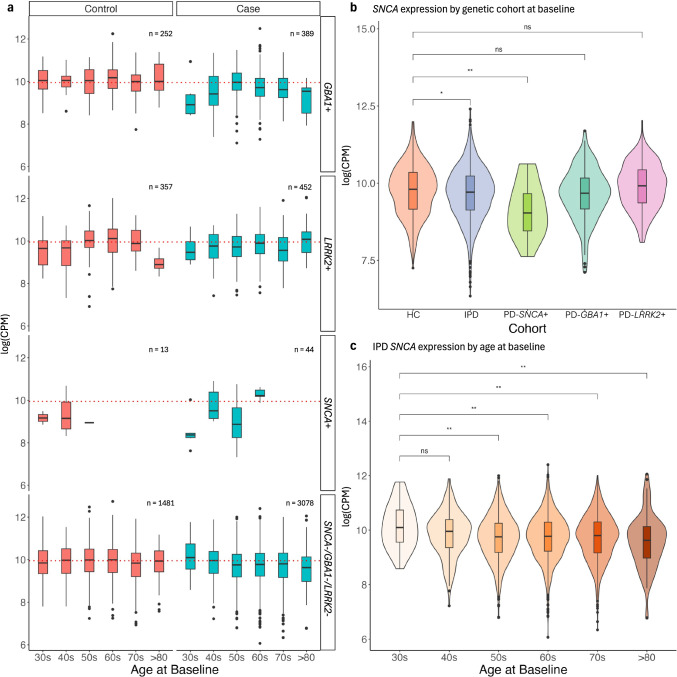
Blood-based RNA transcriptomics offers a promising avenue for identifying biomarkers of Parkinson's disease (PD) progression and mechanisms of pathogenesis. Previous work uncovered an age-related increase of neutrophil-enriched gene expression in PD whole blood, which may obscure disease-relevant transcriptomic signals. To better capture PD-associated molecular differences, we analyzed PD whole-blood RNA sequencing data using a differential expression approach that accounts for neutrophil composition. We built a model to estimate neutrophil percentages in 6897 Parkinson's Progression Markers Initiative and Parkinson's Disease Biomarkers Program samples from gene expression. By incorporating predicted neutrophil percentages as a covariate, we see significant SNCA downregulation in all PD cohorts, a signal previously obscured by immune cell-related effects. Lowered SNCA expression was observed in individuals with known PD-linked gene mutations (e.g., SNCA, GBA1, LRRK2) and those without known pathogenic variants. These findings suggest that decreased SNCA expression in whole blood may be a defining transcriptomic feature of PD.
© 2025. The Author(s).
Conflict of interest statement
Competing interests: The authors declare no competing interests.
Figures
Update of
-
Decreased SNCA Expression in Whole-Blood RNA Analysis of Parkinson's Disease Adjusting for Lymphocytes.bioRxiv [Preprint]. 2024 Nov 19:2024.11.18.623684. doi: 10.1101/2024.11.18.623684. bioRxiv. 2024. Update in: NPJ Parkinsons Dis. 2025 Oct 14;11(1):292. doi: 10.1038/s41531-025-01062-4. PMID: 39605721 Free PMC article. Updated. Preprint.
References
-
- Rizzo, G. et al. Accuracy of clinical diagnosis of Parkinson disease: a systematic review and meta-analysis. Neurology86, 566–576 (2016). - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
