

TET2 repression contributes to EGFR TKI resistance in EGFR-mutant non-small cell lung cancer through regulating AXIN2 methylation
- PMID: 41107309
- PMCID: PMC12534427
- DOI: 10.1038/s41598-025-20263-8
TET2 repression contributes to EGFR TKI resistance in EGFR-mutant non-small cell lung cancer through regulating AXIN2 methylation
Abstract
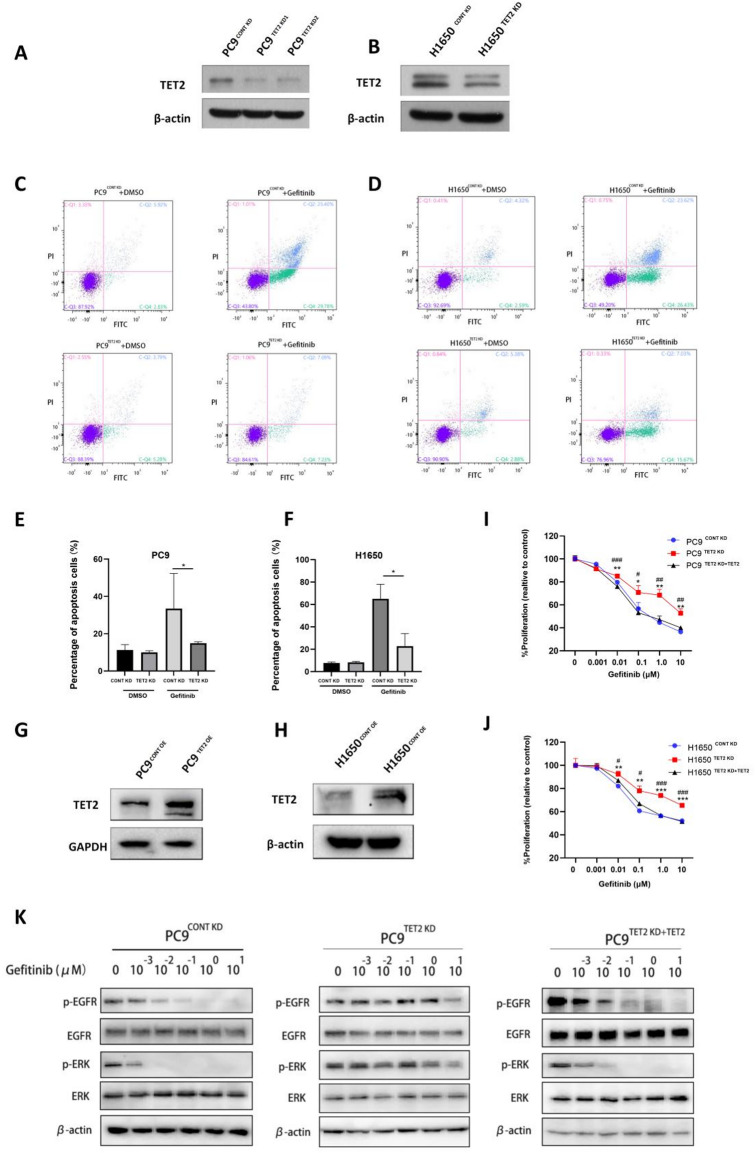
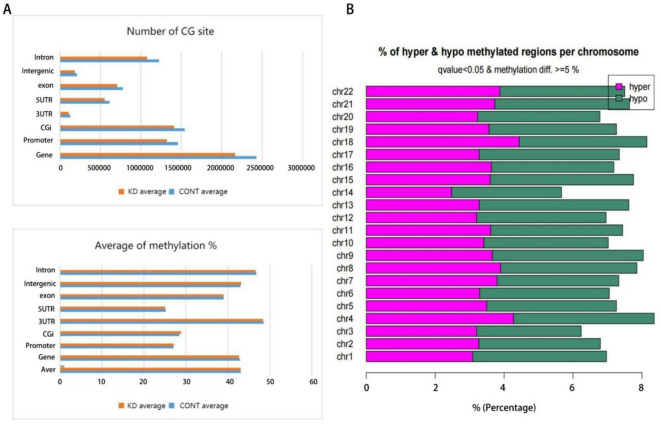
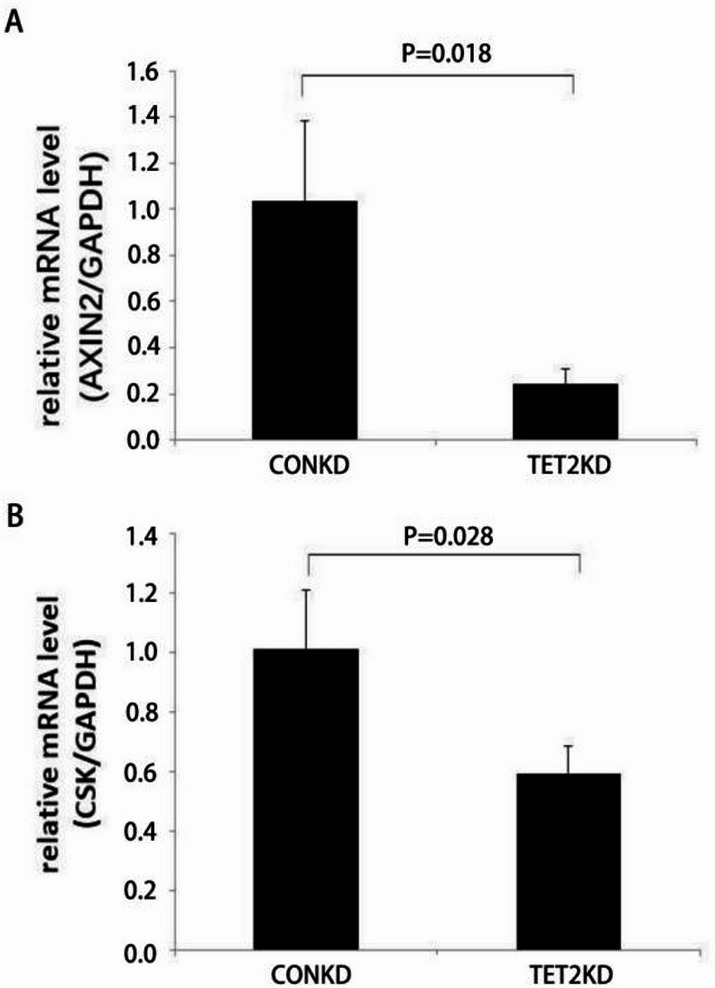
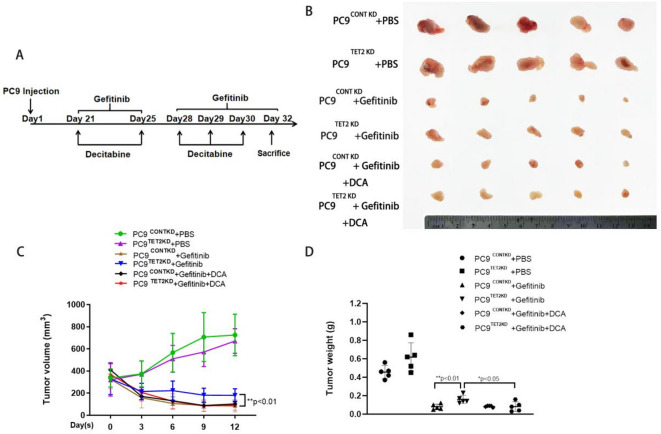
Through targeted next-generation sequencing of 83 non-small cell lung cancer (NSCLC) patients with first-generation epidermal growth factor receptor tyrosine kinase inhibitor (EGFR TKI) resistance, we detected 11% TET2 mutations in the T790M-negative subgroup. To explore the molecular mechanism of TET2 in EGFR TKI resistance, reduced representation bisulfite sequencing (RRBS) was adopted to analyze the global genomic methylation profiles and detect the differentially methylated genes in the TET2-knockdown (KD) PC9 and control PC9 cell lines, following bioinformatics analysis of gene ontology (GO) functions and kyoto encyclopedia of genes and genomes (KEGG) signaling to screen for genes associated with drug resistance. TET2 KD attenuated gefitinib-induced apoptosis and decreased the sensitivity of EGFR-mutant lung cancer cells to gefitinib. Forty-three drug resistance genes with hypermethylated promoter regions were identified via RRBS and bioinformatic analysis in PC9TET2 KD cells. Then, 10 candidate genes were screened for further validation. RT‒PCR demonstrated that the expression of AXIN2 and CSK was significantly lower in PC9TET2 KD cells than in control cells. Furthermore, AXIN2 KD attenuated gefitinib-induced apoptosis and decreased the sensitivity of PC9 cells to gefitinib. Importantly, we found that the demethylation drug decitabine (DCA) could reverse gefitinib resistance in PC9TET2 KD cells and mouse models. These results indicate that the methylation of AXIN2 induced by TET2 repression is a novel resistance mechanism of EGFR TKIs in EGFR-mutant NSCLC. Demethylation drugs have the potential to overcome EGFR TKI resistance induced by loss-of-function TET2 mutations.
Keywords: AXIN2; DNA methylation; EGFR TKI resistance; EGFR-mutant non-small cell lung cancer; TET2 repression.
© 2025. The Author(s).
Conflict of interest statement
Declarations. Competing interests: The authors declare no competing interests.
Figures


References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
