

Forty new genomes shed light on sexual reproduction and the origin of tetraploidy in Microsporidia
- PMID: 41118362
- PMCID: PMC12558613
- DOI: 10.1371/journal.pbio.3003446
Forty new genomes shed light on sexual reproduction and the origin of tetraploidy in Microsporidia
Abstract
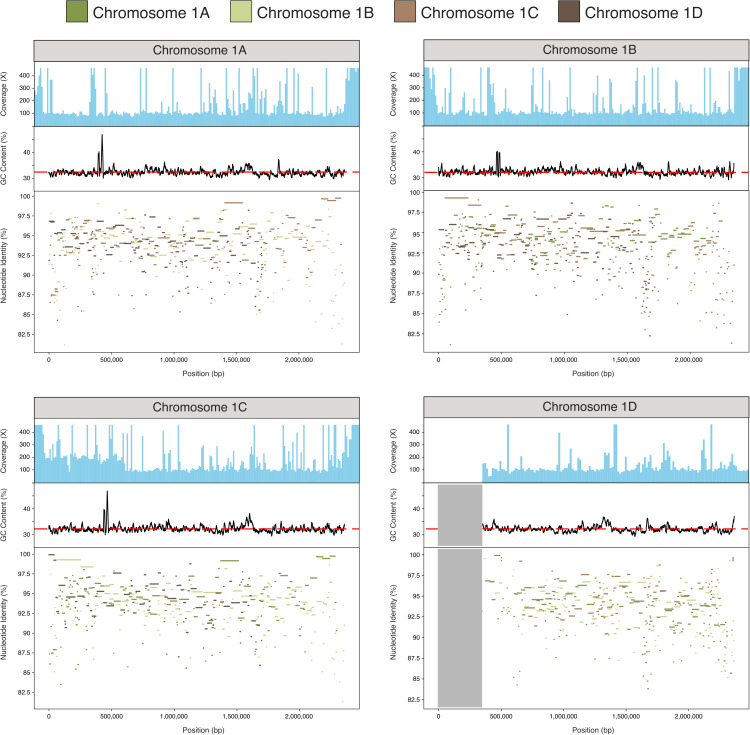
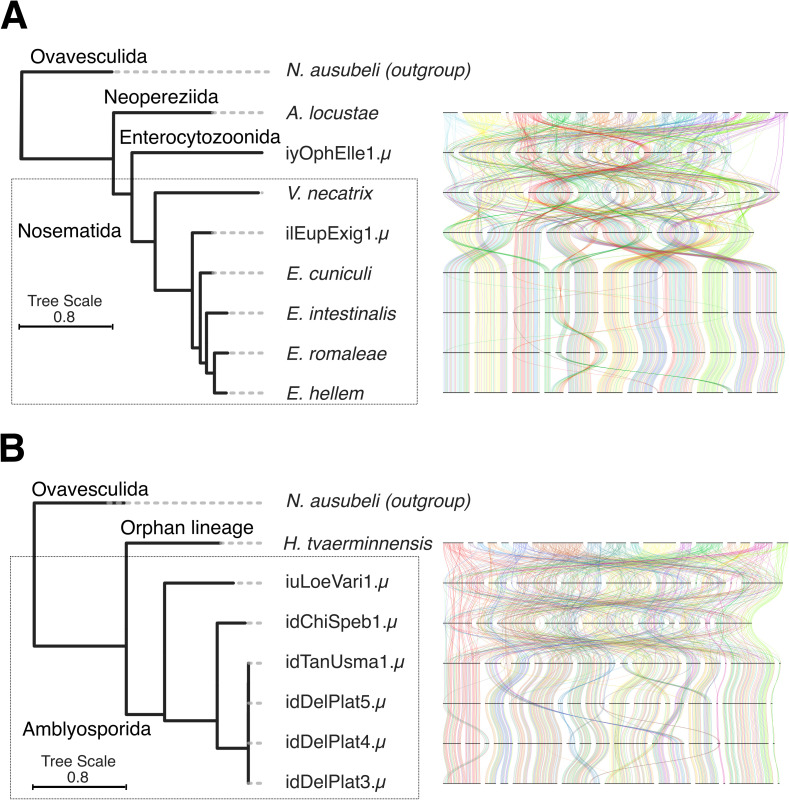
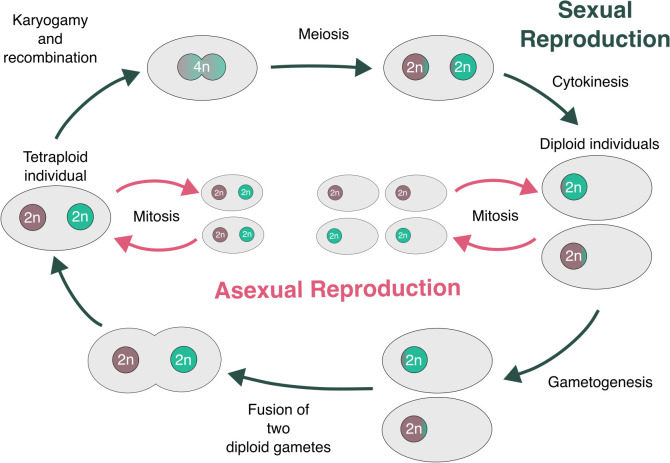
Microsporidia are single-celled, obligately intracellular parasites with growing public health, agricultural, and economic importance. Despite this, Microsporidia remain relatively enigmatic, with many aspects of their biology and evolution unexplored. Key questions include whether Microsporidia undergo sexual reproduction, and the nature of the relationship between tetraploid and diploid lineages. While few high-quality microsporidian genomes currently exist to help answer such questions, large-scale biodiversity genomics initiatives, such as the Darwin Tree of Life project, can generate high-quality genome assemblies for microsporidian parasites when sequencing infected host species. Here, we present 40 new microsporidian genome assemblies from infected arthropod hosts that were sequenced to create reference genomes. Out of the 40, 32 are complete genomes, eight of which are chromosome-level, and eight are partial microsporidian genomes. We characterized 14 of these as polyploid and five as diploid. We found that tetraploid genome haplotypes are consistent with autopolyploidy, in that they coalesce more recently than species, and that they likely recombine. Within some genomes, we found large-scale rearrangements between the homeologous genomes. We also observed a high rate of rearrangement between genomes from different microsporidian groups, and a striking tolerance for segmental duplications. Analysis of chromatin conformation capture (Hi-C) data indicated that tetraploid genomes are likely organized into two diploid units, similar to dikaryotic cells in fungi, with evidence of recombination within and between units. Together, our results provide evidence for the existence of a sexual cycle in Microsporidia, and suggest a model for the microsporidian lifecycle that mirrors fungal reproduction.
Copyright: © 2025 Khalaf et al. This is an open access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures







References
-
- Nageli C. uber die neue Krankheit der Seidenraupe und verwandte Organismen. [Abstract of report before 33. Versamml. Deutsch. Naturf. u. Aerzte. Bonn, 21 Sept.]. Bot Ztg. 1857;15: 760–761. Available from: https://cir.nii.ac.jp/crid/1573105974684833920
-
- Pasteur L. Etudes sur la maladie des vers à soie: 2.: Notes et documents. Gauthier-Villars; 1870. Available from: https://play.google.com/store/books/details?id=y-1rmRQoAa4C
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
