

doi: 10.1016/j.ekir.2025.08.010.
eCollection 2025 Oct.
Functional Evaluation of PKD1 Intronic Variants by Minigene Assays
Affiliations
- PMID: 41141524
- PMCID: PMC12546640
- DOI: 10.1016/j.ekir.2025.08.010
Item in Clipboard
Functional Evaluation of PKD1 Intronic Variants by Minigene Assays
Kidney Int Rep.
.
No abstract available
Keywords: PKD1; minigene assay; splicing variants.
Figures

Minigene assay results of NM_001009944.3(PKD1):c.2097+5G>A. (a) Schematic illustration of pSPL3-PKD1 c.2097+5G>A and c.2853+5G>C minigenes. Exons 10 and 11 of PKD1 and flanking introns were cloned into the pSPL3 vector with a wild-type or mutant c.2097+5G>A and c.2853+5G>C between 2 pSPL3 vectors. After reverse transcription, the splicing products were amplified by polymerase chain reaction using vector exon–specific primers, and (b) the c.2097+5G>A variant was visualized by agarose gel electrophoresis. (c) Sanger sequencing confirmed the reverse transcription–polymerase chain reaction products, and the results in intron 10 showed a 2 bp retention. The clinical profile of the patient harboring the PKD1 variant illustrated in this figure includes the following: onset age of 34 years, an eGFR of 88.09 ml/min per 1.73 m2, classification as Mayo class 1D, and no family history of ADPKD. ADPKD, autosomal dominant polycystic kidney disease; eGFR, estimated glomerular filtration rate; EV, empty vector; NC, normal control; P, proband; SA, splice acceptor exon; SD, splice donor exon; TE, TE buffer.
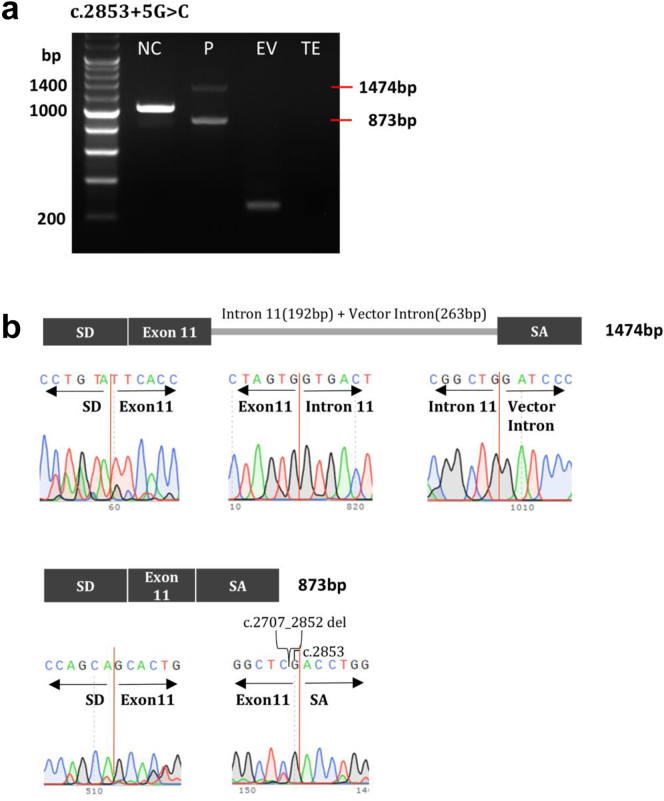
Minigene assay results of NM_001009944.3(PKD1):c.2853+5G>C. (a) HEK293T cells were transfected with wild-type or mutant PKD1 minigene or an empty pSPL3 vector. After reverse transcription, splicing products were amplified by polymerase chain reaction using vector exon–specific primers and (b) visualized by agarose gel electrophoresis. (c) Sanger sequencing confirmed the reverse transcription–polymerase chain reaction products, and the results showed that part of the intron 11 and the vector intron (263 bp) were retained, exon 10 was skipped, and part of exon 11 was deleted because of the presence of a cryptic splicing site. The clinical profile of the patient harboring the PKD1 variant depicted in this figure includes the following: onset age of 44 years, an eGFR of 84 ml/min per 1.73 m2, classification as Mayo class 1C, and a positive family history of ADPKD. ADPKD, autosomal dominant polycystic kidney disease; eGFR, estimated glomerular filtration rate; EV, empty vector; NC, normal control; P, proband; SA, splice acceptor exon; SD, splice donor exon; TE, Tris-EDTA buffer.
References
LinkOut - more resources
Full Text Sources
