

Improved reconstruction of single-cell developmental potential with CytoTRACE 2
- PMID: 41145665
- PMCID: PMC12615260
- DOI: 10.1038/s41592-025-02857-2
Improved reconstruction of single-cell developmental potential with CytoTRACE 2
Abstract
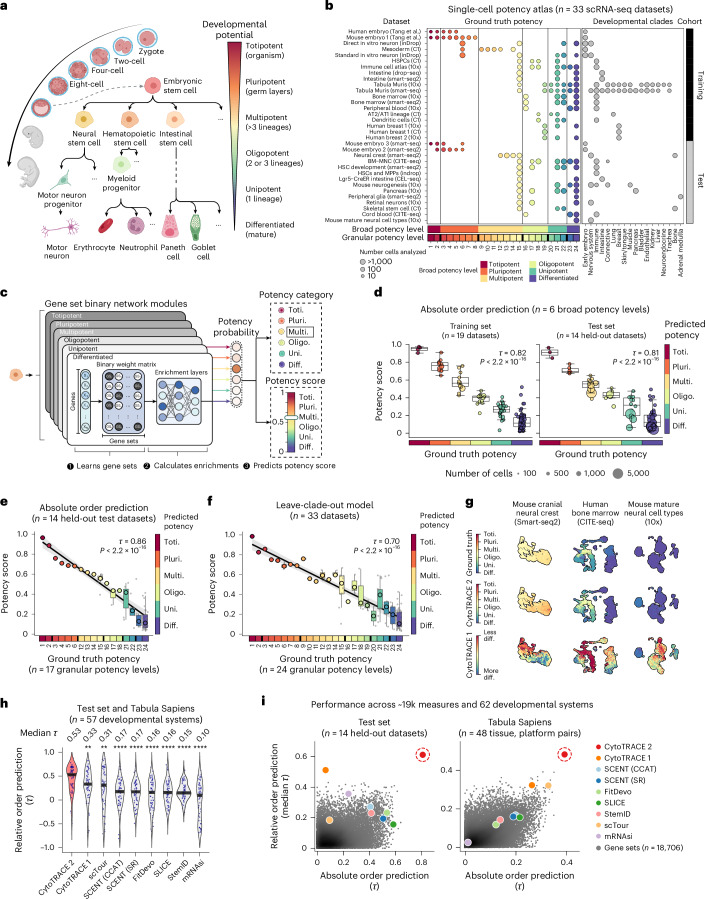
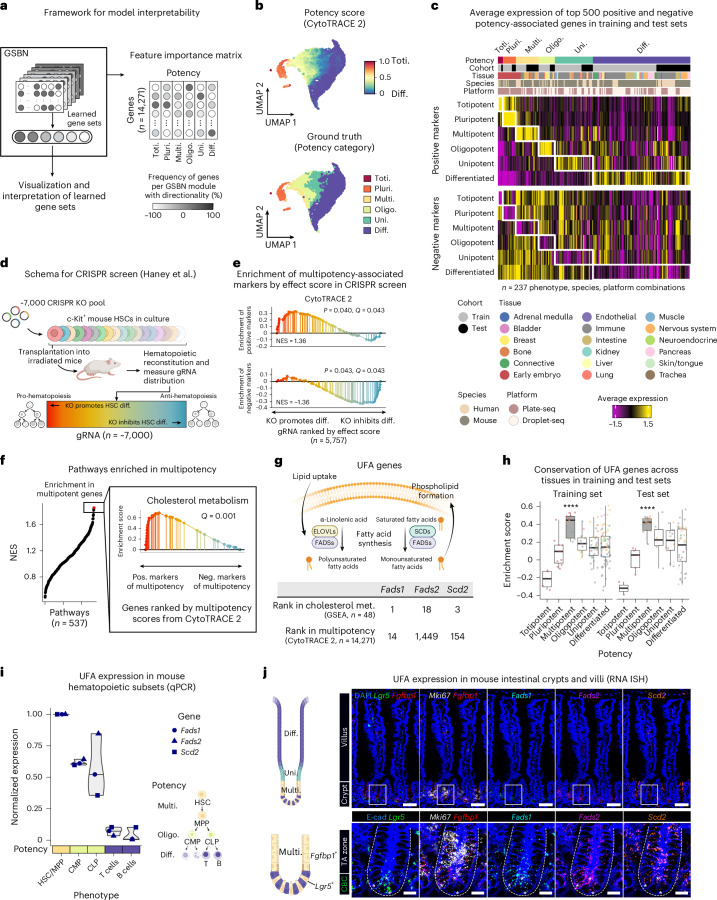
While single-cell RNA sequencing has advanced our understanding of cell fate, identifying molecular hallmarks of potency-a cell's ability to differentiate into other cell types-remains a challenge. Here we introduce CytoTRACE 2, an interpretable deep learning framework for predicting absolute developmental potential from single-cell RNA sequencing data. Across diverse platforms and tissues, CytoTRACE 2 outperformed previous methods in predicting developmental hierarchies, enabling detailed mapping of single-cell differentiation landscapes and expanding insights into cell potency.
© 2025. The Author(s).
Conflict of interest statement
Competing interests: M.K., G.S.G., E.B., J.J.A.A. and A.M.N. have a patent application (US PCT/US25/18429) related to the identification of developmental states from scRNA-seq data. A.M.N. has ownership interests in CiberMed and LiquidCell Dx. A.A.C. has ownership interests in Droplet Biosciences and LiquidCell Dx. All other authors declare no competing interests.
Figures










Update of
-
Mapping single-cell developmental potential in health and disease with interpretable deep learning.bioRxiv [Preprint]. 2024 Mar 21:2024.03.19.585637. doi: 10.1101/2024.03.19.585637. bioRxiv. 2024. Update in: Nat Methods. 2025 Nov;22(11):2258-2263. doi: 10.1038/s41592-025-02857-2. PMID: 38562882 Free PMC article. Updated. Preprint.
References
-
- Bergen, V., Lange, M., Peidli, S., Wolf, F. A. & Theis, F. J. Generalizing RNA velocity to transient cell states through dynamical modeling. Nat. Biotechnol.38, 1408–1414 (2020). - PubMed
MeSH terms
Grants and funding
- R01 CA283317/CA/NCI NIH HHS/United States
- R01CA283317/U.S. Department of Health & Human Services | NIH | National Cancer Institute (NCI)
- R01 CA255450/CA/NCI NIH HHS/United States
- R01CA255450/U.S. Department of Health & Human Services | NIH | National Cancer Institute (NCI)
- 334328/Kirke-, Utdannings- og Forskningsdepartementet (Norwegian Ministry of Education, Research and Church Affairs)
LinkOut - more resources
Full Text Sources
