

Efficacy and safety of risdiplam in adults with 5q-associated spinal muscular atrophy: a nationwide observational cohort study in Austria
- PMID: 41181835
- PMCID: PMC12572788
- DOI: 10.1016/j.eclinm.2025.103536
Efficacy and safety of risdiplam in adults with 5q-associated spinal muscular atrophy: a nationwide observational cohort study in Austria
Abstract
Background: Spinal muscular atrophy (SMA) is a genetic motor neuron disease marked by the progressive decline of motor function. Risdiplam, an orally administered SMN2 splicing modifier, was approved for the treatment of 5q-associated SMA (5q-SMA) across all age groups. However, clinical trial data have primarily focused on paediatric populations, with limited evidence available for adult patients. This study aimed to evaluate the efficacy and safety of risdiplam in treatment-naïve adults with 5q-SMA in a real-world, multicentre setting.
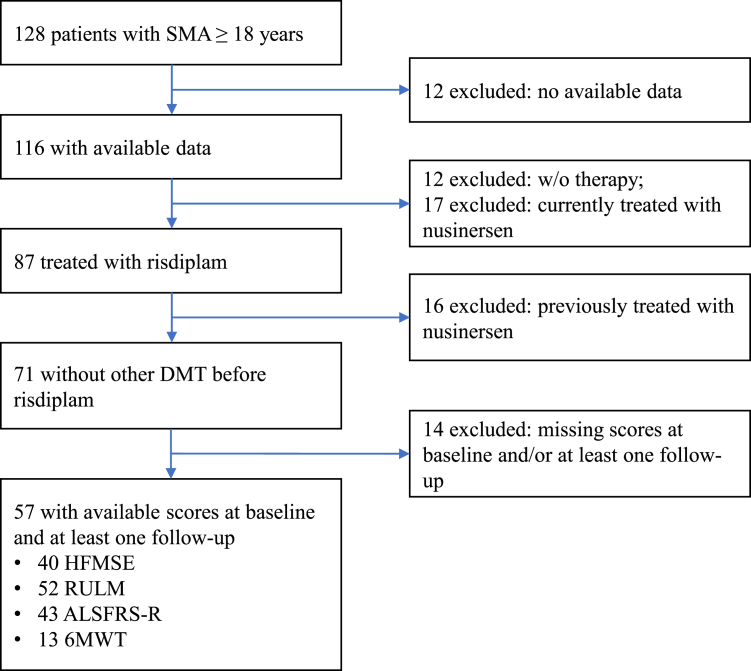
Methods: We conducted a nationwide, observational cohort study across eight neuromuscular centres in Austria. Patients aged ≥16 years at treatment initiation with genetically confirmed 5q-SMA, who were previously untreated and initiated risdiplam between December 2020 and September 2024 were eligible for inclusion if they had received risdiplam for ≥3 months and had functional motor assessments available at baseline (T0) and at least one follow-up. Functional outcomes were assessed at four predefined intervals after baseline: 3-<6 months (T1), 6-<12 months (T2), 12-<18 months (T3), and ≥18 months (T4). The primary outcome was the change from baseline in the Hammersmith Functional Motor Scale Expanded (HFMSE). Secondary outcomes included changes in the Revised Upper Limb Module (RULM), Amyotrophic Lateral Sclerosis Functional Rating Scale-Revised (ALSFRS-R), and 6-min walk test (6MWT). Adverse events were extracted from medical records.
Findings: A total of 87 patients had received risdiplam, of whom 57 fulfilled the inclusion criteria and were included in this study. The median age at treatment initiation was 35.7 years (IQR 28.8-43.4), with a median disease duration of 29.6 years (IQR 24.2-36.3). Most individuals had SMA type II (40.4%) or III (47.4%). Mean HFMSE changes from baseline were +1.00 (95% CI 0.05-1.95, p = 0.0100) at T1, +0.97 (95% CI 0.22-1.72, p = 0.0132) at T2, +1.78 (0.66-2.89, p = 0.0008) at T3, and +1.73 (0.49-2.97, p = 0.0049) at T4. Clinically meaningful improvements in motor function (≥3 points in HFMSE and/or ≥2 in RULM) were observed in 63.9% of patients at T4. Improvements were more pronounced in patients with higher baseline function, ambulatory status, or without a history of spinal surgery. Risdiplam was generally well tolerated, with predominantly mild and non-specific adverse events reported in 14.0% of patients.
Interpretation: In this nationwide observational study in a real-world setting, adult patients with 5q-SMA demonstrated consistent and clinically meaningful functional improvements with risdiplam over time, particularly by 18 months and beyond. These findings support the long-term use of risdiplam in adults with SMA and help close a critical evidence gap in this underrepresented population.
Funding: This study was financially supported by F. Hoffmann-La Roche Ltd.
Keywords: Observational study; Risdiplam; SMA; Spinal muscular atrophy.
© 2025 The Author(s).
Conflict of interest statement
OK received support to attend scientific conferences or meetings from Roche and Biogen and speaker's honoraria from Biogen. ME received honoraria for lectures from Roche and for advisory boards from Roche, ScholarRock and Biogen, as well as support to attend a scientific conference from Roche. MK received travel support from Roche to attend scientific conferences. CK received support to attend scientific meetings as well as travel fees from Roche. TAG received travel compensation and compensation to attend scientific meetings from Biogen and Roche. CGH received support to attend scientific meetings from Biogen, Amicus Therapeutics, and speakers's honoraria from Grünenthal. JT received travel support from Roche to attend scientific conferences. SG received fees for advisory boards from Roche. PM received compensation from Roche to attend an advisory board, and support to attend a scientific meeting from Lundbeck Pharma. MR received consultation fees from Roche. AW received speaker's honoraria as well as conference and travel fees from Roche. MGT received advisory board fees and speaker's honoraria from Novartis and Roche, and support to attend scientific conferences from Roche. SM received consultation fees, speaker's or advisory board honoraria from Novartis, Roche and ScholarRock, and support for attending scientific conferences from Roche. GB received fees for advisory boards and consultations, and support to attend scientific conferences from Novartis, Biogen and Roche. MB received compensation for advisory boards and speaker's honoraria from Novartis, Biogen and Roche and travel compensation to meetings from Roche. RT received support to attend scientific meetings from Angelini, AstraZeneca, Janssen-Cilag, Merz, Pfizer, Sanofi, Biogen and Roche, speaker's honoraria from Abbvie, Alexion, ArgenX, BMS, Daiichi-Sankyo and Teva-ratiopharm. as well as advisory board fees from Alexion, ArgenX, Grünenthal, Janssen-Cilag, Sanofi, UCB, Biogen, Novartis and Roche. CE received fees for advisory boards from Roche and Biogen, and owns stock options from Roche. WL received fees for advisory boards or speaker's honoraria from Novartis, Biogen and Roche, and payments for patents from Roche and Novartis. WL also owns stock options from Novartis. HC received fees for advisory boards and speaker's honoraria from Biogen, Novartis and Roche, and also received funding for research projects from these companies. All other authors declared no conflicts of interest.
Figures


References
-
- Lefebvre S., Burglen L., Reboullet S., et al. Identification and characterization of a spinal muscular atrophy-determining gene. Cell. 1995;80(1):155–165. - PubMed
-
- Battaglia G., Princivalle A., Forti F., Lizier C., Zeviani M. Expression of the SMN gene, the spinal muscular atrophy determining gene, in the mammalian central nervous system. Hum Mol Genet. 1997;6(11):1961–1971. - PubMed
-
- Gregoretti C., Ottonello G., Chiarini Testa M.B., et al. Survival of patients with spinal muscular atrophy type 1. Pediatrics. 2013;131(5):e1509–e1514. - PubMed
-
- Chung B.H., Wong V.C., Ip P. Spinal muscular atrophy: survival pattern and functional status. Pediatrics. 2004;114(5):e548–e553. - PubMed
LinkOut - more resources
Full Text Sources
Miscellaneous
