

Application of error-corrected sequencing technologies for in vivo regulatory mutagenicity assessment
- PMID: 41213378
- PMCID: PMC12645987
- DOI: 10.1016/j.yrtph.2025.105985
Application of error-corrected sequencing technologies for in vivo regulatory mutagenicity assessment
Abstract
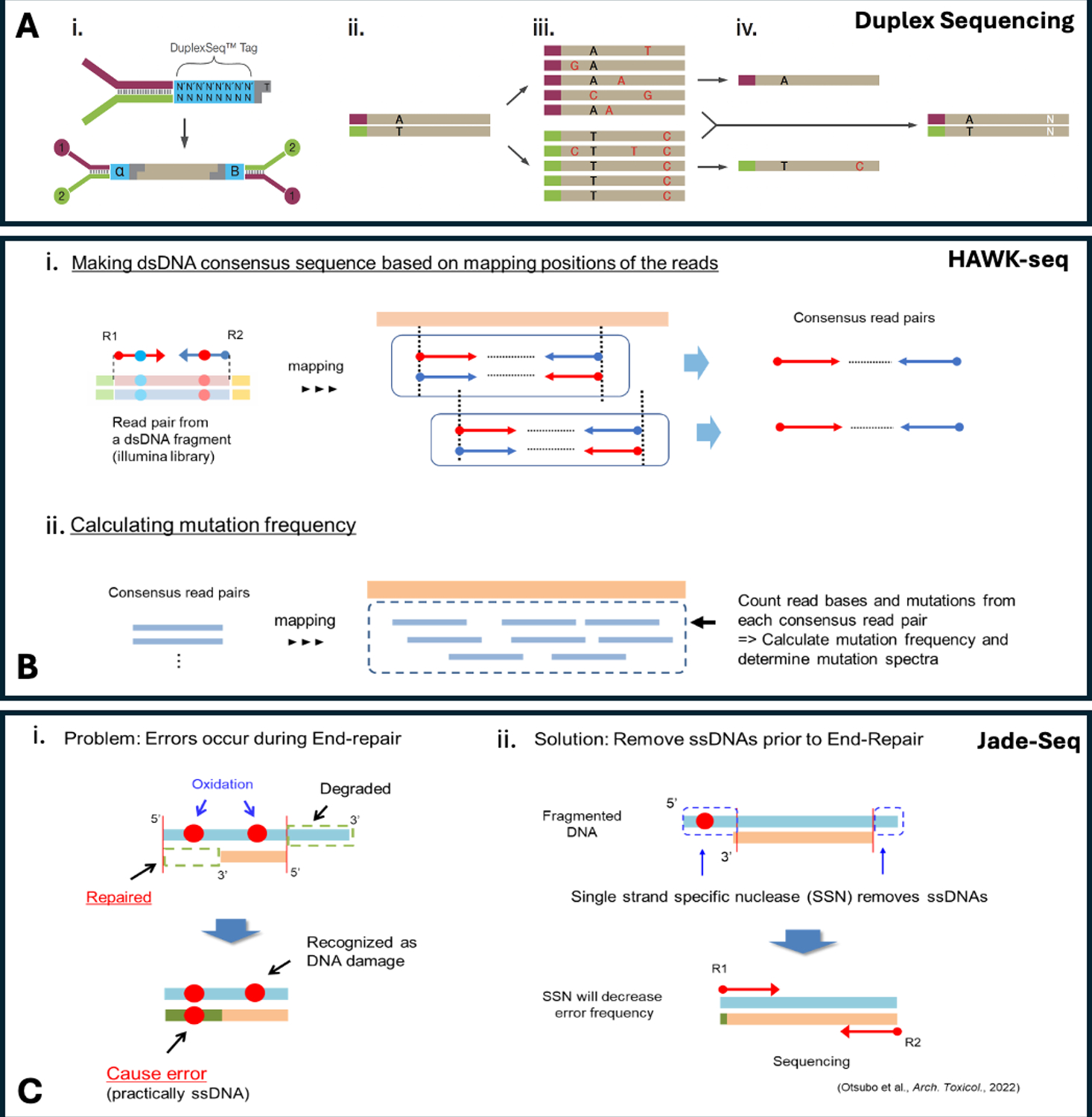
Error-corrected sequencing (ECS) is a transformative method for in vivo mutagenicity assessment, enabling direct, highly sensitive measurement of mutation frequency and spectrum. ECS addresses key limitations of the transgenic rodent (TGR) assay, including lack of integration into standard toxicity studies, restricted model availability, and limited alignment with the 3R principles. To support regulatory acceptance, an expert workgroup of the International Workshops on Genotoxicity Testing (IWGT) reviewed ECS technologies and developed consensus recommendations for its inclusion into Organisation for Economic Co-operation and Development (OECD) test guidelines. The working group agreed that ECS: produces results that are concordant with validated TGR assays; can be incorporated into standard ≥28-day repeat-dose toxicity studies; and, data interpretation should be based on overall mutation frequency compared with concurrent vehicle controls. The working group emphasized harmonized data reporting aligned with OECD principles and endorsed study designs that enable quantitative risk assessment. Overall, the working group agreed that ECS offers a significant advancement over current mutagenicity assays by enabling the use of diverse models beyond conventional TGR systems described in OECD test guideline 488. The working group fully supports the application of ECS to generate in vivo mutagenicity data for regulatory submissions and recommends its inclusion in future OECD test guidelines.
Keywords: Duplex sequencing; Hawk-seq; HiFi-seq; Jade-seq; Mutation; PECC sequencing; SMM-Seq.
Copyright © 2025 The Authors. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of competing interest Jesse J. Salk is a founder, former employee and minority equity-holder of TwinStrand Biosciences Inc. He is a named author on Duplex Sequencing-related patents owned by TwinStrand. He is a named author on Duplex Sequencing patents owned by the University of Washington and licensed to TwinStrand, for which he receives royalties. Devon Fitzgerald is a former employee and equity holder of TwinStrand. She is a named inventor on a pending patent related to Duplex Sequencing for which she is not expected to gain financial benefits. Jake Higgins is equity holder and former employee of TwinStrand. Shoji Matsumura is an employee of Kao Corporation that has applied for the patent for Hawk-Seq™ and Jade-Seq™. Naveed Honarvar is an Editorial Board member of this journal. All other authors declare no conflict of interest.
Figures

References
-
- Abascal F, et al. , 2021. Somatic mutation landscapes at single-molecule resolution. Nature. 593, 405–410. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
