

This is a preprint.
Population-scale Long-read Sequencing in the All of Us Research Program
- PMID: 41256123
- PMCID: PMC12622093
- DOI: 10.1101/2025.10.02.25336942
Population-scale Long-read Sequencing in the All of Us Research Program
Abstract
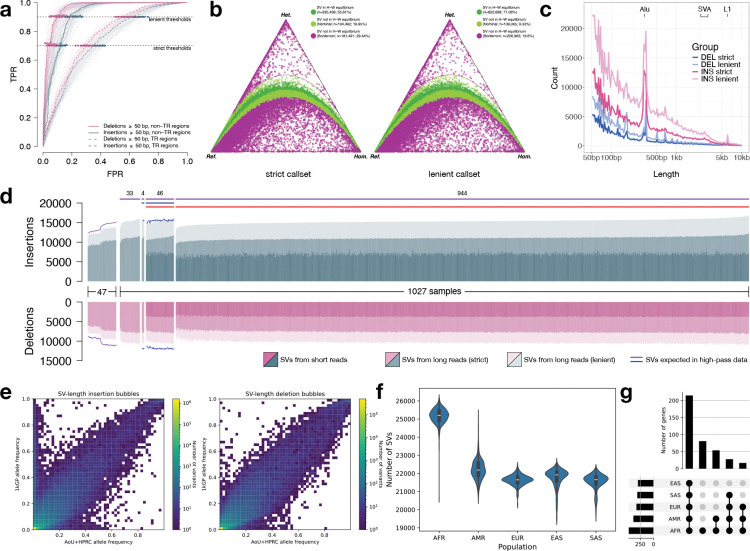
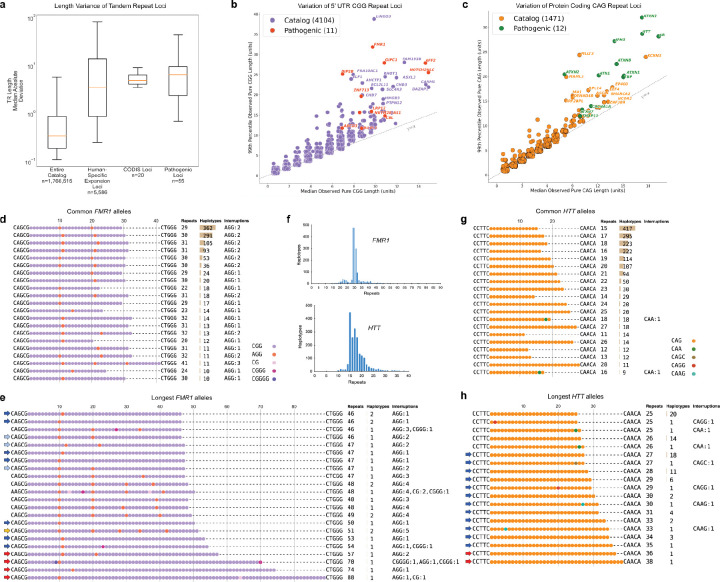
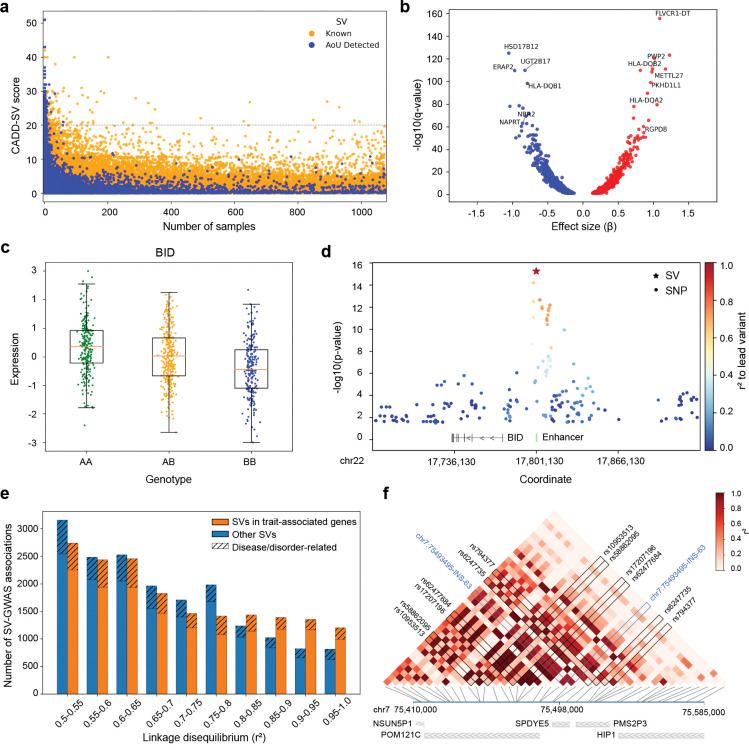
The All of Us Research Program (AoU) is a national biobank seeking to enroll one million individuals in the United States to link genomic and biomedical data, including short- and long-read whole-genome sequencing (srWGS/LRS), with rich electronic health record (EHR) information. Here, we present the first large-scale analyses of long-read sequencing (LRS) in AoU and offer a new framework for deriving genomic insights into complex structural variation (SV) of relevance to human health and disease. We performed joint analyses of 1,027 individuals self-identifying as Black or African American, sequenced to ~8x coverage with Pacific Biosciences HiFi technology and processed using cloud-native pipelines. From these LRS data we constructed a comprehensive variant callset encompassing known (FMR1 and HTT) and novel repeat expansions, clinically relevant haplotypes at loci inaccessible to srWGS, and haplotypes relevant to disease risk (HLA) and pharmacogenomics (CYP2D6), including SNVs, indels, and SVs. We developed methods for cohort-level variant calling and a scalable workflow to impute >750,000 of these SVs into existing srWGS datasets for trait association and human disease studies. Expanding to 10,000 self-identified Black or African American AoU participants with srWGS and matched EHRs, we identified 291 SV-disease associations (p < 1×10-5) spanning 226 conditions with 50.9% of associations involving SVs absent from the matched srWGS callset. Across the 226 traits, after fine-mapping using SVs and SNVs we identified 191 SV-disease pairs spanning 160 traits (70.8%) where the SV had the strongest association within the locus. Associations specific to those with computed ancestry similar to the African reference population exhibited larger effect sizes and lower allele frequencies, consistent with high-risk, ancestry-specific variants. These results demonstrate that the integration of LRS into AoU and future biobank initiatives can provide transformative new insights into genomic variation with potentially profound impact on precision medicine.
Conflict of interest statement
Ethics declarations / Competing Interests K.V.G. is a co-inventor on a pending international patent application related to long-read RNA isoform sequencing, licensed to Pacific Biosciences, but not used in this study. E.E.E. is a scientific advisory board (SAB) member of Variant Bio, Inc. F.J.S. receives research support from Illumina and Nanopore. The remaining authors declare no competing interests.
Figures



References
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials