

This is a preprint.
Host transcriptional responses to gut microbiome variation arising from urbanism
- PMID: 41278623
- PMCID: PMC12633242
- DOI: 10.1101/2025.10.26.683539
Host transcriptional responses to gut microbiome variation arising from urbanism
Abstract
Gut microbiomes of urban communities are compositionally different from their rural counterparts, and are associated with immune dysregulation and gastrointestinal disease. However, it is unknown whether these compositional differences impact host physiology, and through what mechanisms. Here, we used human colonic epithelial cells to directly compare host transcriptional changes induced by gut microbiomes from urban versus rural communities. We co-cultured host cells with live, stool-derived gut microbiomes from Rwanda, Ghana, Nigeria, Malaysia, and the United States, and quantified transcriptional responses using RNA-seq. We found that urban microbiomes affected innate immune pathways, including TNF signaling and bacterial antigen recognition. We also found that high-diversity microbiomes elicited a stronger host transcriptional response, while low-diversity microbiomes triggered epithelial restructuring and glycolysis. Finally, specific taxa driving these effects, including Bifidobacterium adolescentis and Bacteroides dorei, correlated with lifestyle factors such as diet. These findings demonstrate that urbanization-associated microbiome changes directly influence host epithelial gene expression.
Conflict of interest statement
Competing interests The authors declare no competing interests.
Figures
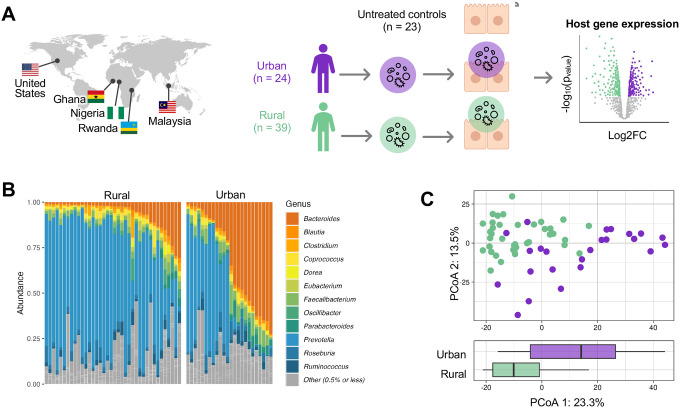

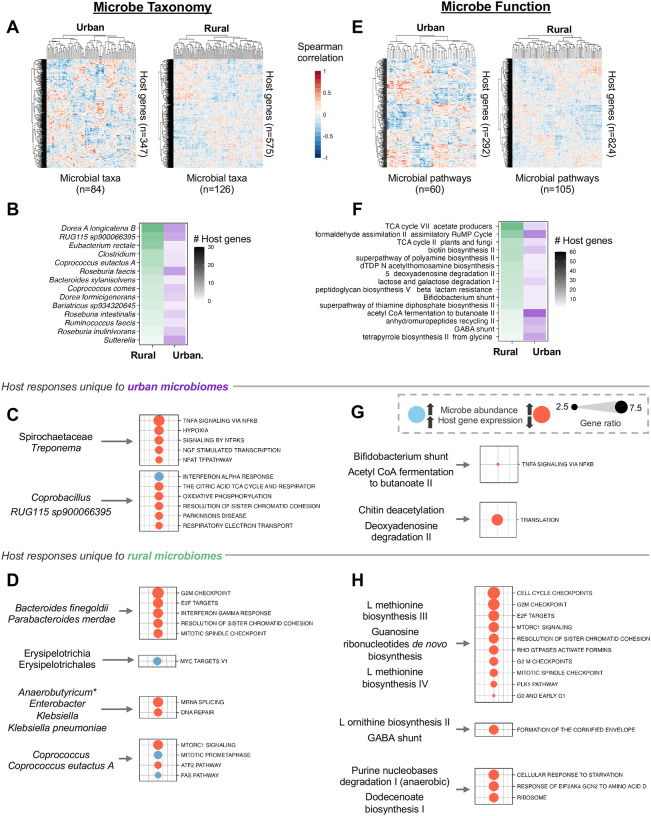

References
-
- Shanahan F, Ghosh TS, O’Toole PW. The healthy microbiome-what is the definition of a healthy gut microbiome? Gastroenterology. 2021;160:483–94. - PubMed
-
- Parizadeh M, Arrieta M-C. The global human gut microbiome: genes, lifestyles, and diet. Trends Mol Med. 2023;29:789–801. - PubMed
-
- Abdill RJ, Graham SP, Rubinetti V, Ahmadian M, Hicks P, Chetty A, et al. Integration of 168,000 samples reveals global patterns of the human gut microbiome. Cell [Internet]. 2025. [cited 2025 Feb 6];0. Available from: http://www.cell.com/article/S0092867424014302/abstract
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources