

This is a preprint.
Industrialization drives convergent microbial and physiological shifts in the human metaorganism
- PMID: 41280098
- PMCID: PMC12633315
- DOI: 10.1101/2025.10.20.683358
Industrialization drives convergent microbial and physiological shifts in the human metaorganism
Abstract
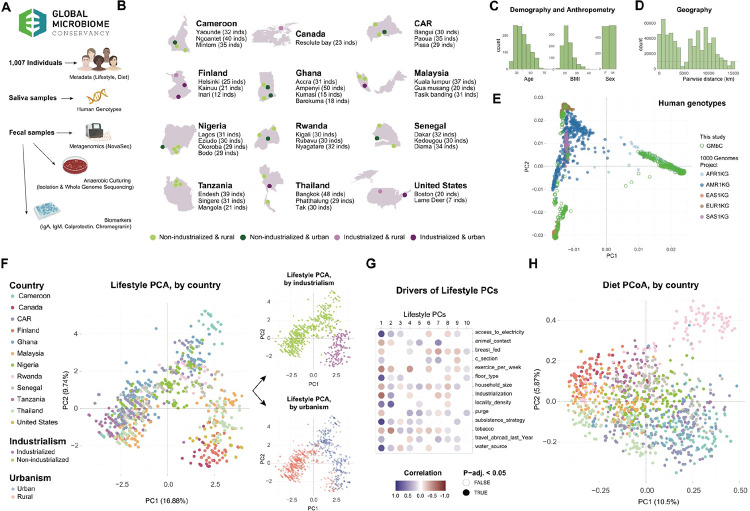
Understanding how host lifestyle and industrialization shape the human gut microbiome and intestinal physiology requires multimodal analyses across diverse global host contexts. Here, we generate multivariate data from the Global Microbiome Conservancy cohort, including gut microbiome, IgA-sequencing, host genotyping, diet, lifestyle and fecal biomarker profiles, to investigate host-microbiome interactions across gradients of industrialization and geography. We show that industrialization is associated with homogenized microbial compositions, reduced microbial diversity, and lower community stability, independent of host confounders. We further show that industrialization is linked to elevated markers of gut stress, increased IgA secretion, and altered patterns of IgA-bacteria interactions. Finally, we show that microbiome-based disease predictors trained on industrialized populations lose accuracy in less industrialized cohorts, highlighting limited cross-population transferability. Together, our results suggest profound restructuring of host-microbiome interactions due to industrialized lifestyles, and emphasize the need for inclusive, globally representative data to improve translational microbiome applications across diverse human populations.
Conflict of interest statement
R.J.X. is a co-founder of Convergence Bio, board director at MoonLake Immunotherapeutics, a consultant to Nestlé, and a member of the advisory boards at MagnetBiomedicine and Arena Bioworks. R.K. is a scientific advisory board member and consultant for BiomeSense, Inc., has equity, and receives income. He is a scientific advisory board member and has equity in GenCirq. He is a consultant and scientific advisory board member for DayTwo and receives income. He has equity in and acts as a consultant for Cybele. He is a co-founder of Biota, Inc., and has equity. He is a co-founder of Micronoma and has equity and is a scientific advisory board member. The terms of these arrangements have been reviewed and approved by the University of California, San Diego, in accordance with its conflict of interest policies. D.M. is a consultant for and has equity in BiomeSense, Inc. The terms of these arrangements have been reviewed and approved by the University of California, San Diego, in accordance with its conflict-of-interest policies. No organizations listed above provided funding for this study.
Figures






References
-
- Ayeni F.A., Biagi E., Rampelli S., Fiori J., Soverini M., Audu H.J., Cristino S., Caporali L., Schnorr S.L., Carelli V., et al. (2018). Infant and Adult Gut Microbiome and Metabolome in Rural Bassa and Urban Settlers from Nigeria. Cell Rep. 23, 3056–3067. 10.1016/j.celrep.2018.05.018. - DOI - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous